
Abstract
The purpose of this study was to investigate the safety, tolerability, and pharmacokinetics of motesanib when combined with docetaxel or paclitaxel in patients with metastatic breast cancer. In this open-label, dose-finding, phase 1b study, patients received motesanib 50 or 125-mg orally once daily (QD), beginning day 3 of cycle 1 of chemotherapy, continuously in combination with either paclitaxel 90 mg/m2 on days 1, 8, and 15 every 28-day cycle (Arm A) or docetaxel 100 mg/m2 on day 1 every 21-day cycle (Arm B). Dose escalation to motesanib 125 mg QD occurred if the incidence of dose-limiting toxicities (DLTs, primary endpoint) was ≤33 %. If the maximum tolerated dose (MTD) of motesanib was established in Arm B, additional patients could receive motesanib at the MTD plus docetaxel 75 mg/m2. Forty-six patients were enrolled and 45 received ≥1 dose of motesanib. The incidence of DLTs was <33 % in all cohorts; thus, motesanib 125 mg QD was established as the MTD. Seven patients (16 %) had grade 3 motesanib-related adverse events including cholecystitis (2 patients) and hypertension (2 patients). Pharmacokinetic parameters of motesanib were similar to those reported in previous studies. The objective response rate was 56 % among patients with measurable disease at baseline who received motesanib in combination with taxane-based chemotherapy. The addition of motesanib to either paclitaxel or docetaxel was generally tolerable up to the 125-mg QD dose of motesanib. The objective response rate of 56 % suggests a potential benefit of motesanib in combination with taxane-based chemotherapy.
Similar content being viewed by others


Introduction
Although there has been a significant decline in breast cancer mortality over the last decade, up to 40 % of patients will develop metastatic breast cancer (MBC), for which there remains no curative therapy. Many therapeutic agents effectively treat MBC; however, the overall duration of response remains far from ideal. The median survival from diagnosis for triple negative MBC is approximately 2 to 3 years and for estrogen receptor-positive/progesterone receptor-positive disease, approximately 5 years [1]. Thus, the development of new therapies to treat MBC remains critically important.
Angiogenesis is essential for breast cancer development and metastasis [2], and high tumor levels of the proangiogenic cytokine vascular endothelial growth factor (VEGF) are predictive of poor clinical outcomes in patients with breast cancer [3, 4]. The VEGF signaling pathway has thus become a promising target, and agents targeting this pathway have been shown to improve outcomes in patients with MBC [5].
Motesanib is an orally administered, small-molecule antagonist of VEGF receptors (VEGFR) 1, 2, and 3; platelet-derived growth factor receptor (PDGFR); and Kit [6]. Treatment with motesanib was tolerable and showed antitumor activity when administered as monotherapy to patients with advanced solid tumors [7, 8] and in combination with either chemotherapy or an anti-epidermal growth factor receptor antibody in patients with non-small-cell lung cancer (NSCLC) [9]. In tumor xenograft models of human breast cancer, treatment with motesanib resulted in reductions in tumor growth and tumor blood vessel density [10]. Moreover, additive reductions in tumor growth were achieved when motesanib was combined with docetaxel [10], possibly as a result of VEGF pathway blockade enhancing (and/or conserving) the antiangiogenic activity of the taxane [11]. Potentially, the combination of motesanib with taxane chemotherapy may have activity in patients with MBC. The objective of this phase 1b study was to investigate the safety, tolerability, and pharmacokinetics of motesanib when combined with taxanes (docetaxel or paclitaxel) in patients with MBC.
Materials and methods
Patients
Female patients ≥18 years old were eligible if they had confirmed measurable or nonmeasurable [per Response Evaluation Criteria in Solid Tumors (RECIST) v1.0 [12]] adenocarcinoma of the breast with locally recurrent or metastatic disease, Eastern Cooperative Oncology Group performance status of 0/1, and adequate organ function. Exclusion criteria included >1 prior chemotherapy regimen for MBC; taxane-containing treatment within 6 months, bevacizumab within 3 months, or VEGFR-targeted therapy within 1 month before enrollment; uncontrolled hypertension; prior malignancy (except in situ cervical cancer or nonmelanoma skin cancer); radiation therapy to >25 % of bone marrow; radiation therapy for peripheral lesions within 14 days of enrollment; central nervous system metastases; arterial or venous thrombosis within 12 months before enrollment; bleeding diathesis or bleeding within 14 days or major surgery within 28 days before enrollment; clinically significant cardiac disease; and prior episodes of cholecystitis.
The study protocol was approved by the Institutional Review Board/Ethics Committee at each participating study site, and all patients provided written consent.
Study design
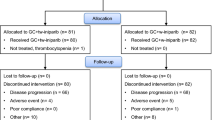
This open-label, dose-finding, multicenter study conducted at 6 centers in Australia and 1 in the United States evaluated the safety of motesanib in combination with paclitaxel or docetaxel. Patients with locally recurrent or metastatic breast cancer were assigned to receive motesanib in combination with paclitaxel (Arm A), or motesanib in combination with docetaxel (Arm B) (Fig. 1). The maximum planned sample size (if all cohorts enrolled the maximum number of patients) was 54. Four dose cohorts (2 in Arm A and 2 in Arm B) that were initially planned to test lower doses of motesanib (100 and 75 mg) were not opened because the 125-mg dose was found to be tolerable. Hence, only 46 patients were ultimately enrolled in the study. The primary endpoint was the incidence of dose-limiting toxicities (DLTs). Secondary endpoints included the incidence of adverse events (AEs); pharmacokinetics of motesanib, paclitaxel, and docetaxel; objective tumor response (per RECIST v1.0) [12]; and duration of response. Evaluation of pharmacodynamic biomarkers was an exploratory endpoint.

Study schema. aThe sponsor and the principal investigators reviewed the safety data from each cohort to evaluate possible drug effects and DLTs. bDisease progression or motesanib intolerability
Maximum tolerated dose (MTD) and DLT
The MTD was defined as the highest dose of motesanib with an observed incidence of DLT in ≤33 % of patients per cohort.
A DLT was defined as any grade 3 or 4 hematologic or nonhematologic toxicity (except alopecia) occurring during cycle 1 that was related to motesanib or the combination of motesanib plus chemotherapy. Fatigue, nausea, diarrhea, vomiting, neutropenia, febrile neutropenia, neuropathy, thrombocytopenia, anemia, hypertension, and aspartate aminotransferase (AST) or alanine aminotransferase (ALT) elevations were only considered DLTs if the following conditions were met: grade 3 fatigue >7 days or grade 4 fatigue; grade 3 or 4 nausea, diarrhea, or vomiting despite maximum supportive care; grade 3 or 4 neutropenia with fever ≥38.5 °C; grade 4 neutropenia (absolute neutrophil count <0.5 × 109/L) for >7 days; grade 4 thrombocytopenia (platelet count <25 × 109/L) for >7 days; grade 4 anemia; grade 4 hypertension; or AST or ALT > 10 times the upper limit of normal.
Administration of study drugs and dose escalation
Patients were assigned to receive motesanib (Amgen Inc., Thousand Oaks, CA) plus either paclitaxel or docetaxel. Motesanib 125 mg administered orally once daily (QD) was the MTD in the first-in-human single-agent motesanib study [7] and was the maximum dose for this study. Patients received 1 of 2 doses of motesanib (50 or 125 mg) self administered orally QD beginning day 3 of cycle 1, and then from day 1 of subsequent cycles (Fig. 1). Motesanib was administered with paclitaxel 90 mg/m2 [intravenously (IV) over 1 h ± 15 min] on days 1, 8, and 15 every 28-day cycle (Arm A) or docetaxel 100 mg/m2 or 75 mg/m2 (IV over 1 h ± 15 min) on day 1 every 21-day cycle (Arm B). Patients receiving motesanib plus docetaxel 100 mg/m2 received myeloid growth factor support as primary prophylaxis for febrile neutropenia.
Up to 6 patients were enrolled in each arm at the starting dose of motesanib 50 mg QD. Dose escalation to motesanib 125 mg QD was allowed if ≥4 patients completed cycle 1 with no DLTs. At least 4 patients could be enrolled into subsequent dosing cohorts, all of which could be expanded to acquire additional safety data. If >2 of 4 patients receiving motesanib 125 mg QD experienced a DLT, lower doses could be explored. If the MTD of motesanib was established in Arm B, an additional cohort of patients could be enrolled to receive motesanib at the MTD plus docetaxel 75 mg/m2, a commonly used dose of docetaxel for patients with MBC in many countries; this cohort received no myeloid growth factor support. Intrapatient dose escalation was not allowed. Patients continued to receive study drugs until disease progression, withdrawal of consent, or occurrence of unacceptable toxicity.
Dose modifications
If a patient experienced a DLT, motesanib was withheld until the toxicity resolved to either grade 1 or baseline and then restarted with a 25-mg dose reduction. One dose reduction per patient was allowed with the exception of hypertension management, for which 2 dose reductions were allowed. If the patient did not recover within 3 weeks, study treatment was discontinued. A DLT in patients receiving motesanib 50 mg QD would result in the discontinuation of study treatment.
Chemotherapy could be delayed ≤3 weeks for severe toxicities. One dose reduction was allowed for paclitaxel; 2 were allowed for docetaxel. More than a 3-week delay in treatment resulted in permanent discontinuation of chemotherapy.
Blood pressure was monitored weekly for the first 6 weeks and then at each clinic visit. Investigators were permitted to use standard antihypertensive treatments. Motesanib was to be discontinued for reoccurrence of symptomatic hypertension or hypertension despite maximal doses of a 4-drug antihypertensive regimen. Motesanib was also permanently discontinued for patients with grade 4 hemorrhage, >1 grade 3 hemorrhage, grade 4 venous thrombosis, or grade 3 or 4 arterial thrombosis.
Motesanib was withheld for patients who developed a clinical diagnosis of cholecystitis or symptoms attributed to gallbladder enlargement in the absence of cholecystitis. Patients who developed hypothyroidism [elevated thyroid-stimulating hormone (TSH) levels above upper limits of normal and/or a low T4 level] and/or signs or symptoms of hypothyroidism could receive thyroid hormone replacement therapy (i.e., levothyroxine) according to standard clinical care.
AE assessments
AEs were recorded and classified according to the Medical Dictionary for Regulatory Activities and graded according to the Common Terminology Criteria for Adverse Events version 3.0 [13].
Pharmacokinetic and pharmacodynamic analyses
Plasma samples for pharmacokinetic analysis of motesanib and analysis of serum placental growth factor (PLGF) and VEGF were collected predose and at 1, 3, 6, 24 (before the next motesanib dose), and 48 h (trough) after motesanib administration on days 3 and 8 of cycle 1 in Arm A and on day 3 of cycle 1 and day 1 of cycle 2 in Arm B. Plasma samples for pharmacokinetic analysis of paclitaxel and docetaxel were collected preinfusion and at 0.5, 1, 2, 4, 6, 24, 48, and 96 h after infusion on days 1 and 8 of cycle 1 in Arm A and on day 1 of cycles 1 and 2 in Arm B. Samples were analyzed at Amgen, Inc. (Thousand Oaks, CA) for motesanib and at Bioanalytical Systems, Inc. (McMinnville, OR) for paclitaxel and docetaxel using validated analytical methods. Serum PLGF and VEGF were assessed using multiplexed electrochemiluminescent immunoassays (Meso-Scale Discovery, Gaithersburg, MD) as previously described [14].
Pharmacokinetic parameters including the area under the concentration–time curve (AUC) and the maximum and minimum observed plasma concentrations (C max and C min) were estimated using standard noncompartmental methods with WinNonlin software (version 5.1.1, Pharsight Corporation, Mountain View, CA). The effect of motesanib on exposure to paclitaxel and docetaxel was investigated by calculating the ratio point estimates for the geometric least square mean (GLSM) values (90 % CI) of C max and AUC0-inf for motesanib plus paclitaxel versus paclitaxel alone (day 8 vs. 1) and motesanib plus docetaxel versus docetaxel alone (cycle 2 vs. 1) using SAS PROC Mixed procedure (SAS for Windows, version 9.1, WIN_PRO platform; SAS Institute, Inc.). Specifically, GLSM ratios were calculated by estimating the difference in the least squares means for log-transformed C max and AUC0-inf; the ratios were then converted back to their original scale.
Tumor-response assessment
Tumor response was assessed with either computed tomography or magnetic resonance imaging within 28 days of enrollment and every 2 cycles: every 8 ± 1 weeks in Arm A and every 6 ± 1 weeks in Arm B. Bone scans were performed every 12 ± 1 weeks if bone metastases were present at baseline and at any time of clinical suspicion in other patients. Tumor response was assessed by investigators per modified RECIST v1.0 [12].
Statistical analyses
Safety was evaluated among all patients who received ≥1 dose of motesanib. Tumor responses were recorded for all patients with measurable disease at baseline. Duration of response was calculated as the time from the first objective response to disease progression or death. Patients who responded and did not progress or die while on study were censored on the date of their last assessment. Progression-free survival (PFS) was calculated as the time from the first day of study treatment and the date when disease progression was determined or death. Patients who had not died and did not have an assessment of disease progression were censored.
Results
Patients
Forty-six patients were enrolled between May 2006 and August 2008. One patient was screened but withdrew consent before receiving study treatment. Forty-five patients received ≥1 dose of motesanib. Patient demographics and baseline characteristics are summarized in Table 1.
Forty-two patients discontinued motesanib because of disease progression (n = 23, 50 %), AE (n = 15, 33 %), administrative decision (n = 2, 4 %), and withdrawal of consent (n = 2, 4 %). Three patients were receiving motesanib at the time of data analysis. Ten of 19 patients in cohort B2 and 8/12 patients in cohort B3 had dose reductions during the study; dose reductions were not required in other cohorts. The median follow-up time was 29.5 weeks (range 1–94 weeks).
DLTs and MTD
Five patients received motesanib 50 mg QD plus paclitaxel 90 mg/m2 (Cohort A1), and 5 received motesanib 50 mg QD plus docetaxel 100 mg/m2 (Cohort B1). None of these patients had a DLT; therefore, 5 patients were enrolled into Cohort A2 and received motesanib 125 mg QD plus paclitaxel 90 mg/m2 and 19 were enrolled into Cohort B2 and received motesanib 125 mg QD plus docetaxel 100 mg/m2. There were no DLTs in Cohort A2; DLTs occurred in 3 patients (16 %) in Cohort B2, 2 with grade 3 fatigue >7 days and 1 with grade 3 migraine. Motesanib 125 mg QD was, therefore, established as the MTD, and 12 additional patients were enrolled into Cohort B3 and received motesanib 125 mg QD plus docetaxel 75 mg/m2; no DLTs occurred in this cohort.
Adverse events
All 45 patients experienced ≥1 treatment-emergent AE during the study, of whom 29 (64 %), 6 (13 %), and 2 (4 %) had grade 3, 4, or 5 events, respectively. Grade 4 AEs included neutropenia (Cohort A2, 1 patient; Cohort B2, 2 patients; and Cohort B3, 1 patient), pneumonia and acute respiratory failure (Cohort B2, 1 patient), and pyrexia (Cohort B3, 1 patient). Grade 5 AEs occurred in 1 patient (Cohort B1) who experienced grade 4 thrombocytopenia, influenza, and neutropenic sepsis and later died of bronchopneumonia; and 1 in Cohort B2 who experienced grade 3 migraine, hypertension, fatigue, diplopia, asthenia, hyperbilirubinemia, and decreased level of consciousness (after a fall) and died of multiorgan failure. None of the grade 4 or 5 AEs were considered related to treatment with motesanib.
Forty patients (89 %) experienced ≥1 motesanib-related treatment-emergent AE, 19 of whom (42 %) had grade 3 motesanib-related AEs, the most common being fatigue and diarrhea (Table 2). The incidence of grade 3 motesanib-related AEs was approximately 2-fold higher among patients who received motesanib 125 mg QD versus 50 mg QD.
Twenty-seven patients (60 %) had treatment-emergent AEs of interest deemed related to motesanib, 7 of whom (16 %) had grade 3 AEs (Table 2). The patients with grade 3 cholecystitis, grade 3 decreased ejection fraction, or grade 3 increased blood amylase were removed from the study. The 2 patients with hypertension had recurrent hypertension (despite medication) and had their dose of motesanib altered. Each of these AEs occurred after the DLT assessment window.
Grade 2 hypertension deemed related to motesanib was observed in 9 patients; 2 had recurrent hypertension, and 1 was removed from the study.
Pharmacokinetics of motesanib, paclitaxel, and docetaxel
Motesanib AUC, C max, and C min values were comparable to those observed in previous studies (Fig. 2). Pharmacokinetic parameters are shown in Table 3. Coadministration of motesanib with paclitaxel increased paclitaxel C max by 23–45 % and increased paclitaxel AUC0-inf by 18–28 %. Coadministration of motesanib with docetaxel did not affect docetaxel C max or docetaxel AUC0-inf in Cohort B2; however, the data in Cohorts B1 and B3 showed high variability (GLSM values ranging from 0.98 to 2.47) due to 1 patient in each cohort with unusually high exposures during Cycle 2. Excluding these patients from the analysis, coadministration of motesanib did not appear to markedly affect docetaxel exposure in Cohort B1, although C max increased 53 % and AUC0-inf decreased 19 % in Cohort B3.

Comparison of motesanib C max (a) and AUC0-inf (b) values during cycle 1 with C max and AUC0-inf values obtained from previous motesanib studies. Study 1 is the first-in-human study of motesanib in patients with advanced solid tumors [7]; study 2 is the phase 1b study of motesanib in combination with chemotherapy or panitumumab in patients with NSCLC [9]
Pharmacodynamic biomarkers
PLGF concentrations in the serum increased 24 h after initiation of motesanib. In all analyzed cohorts, approximately 3-fold increases from baseline in PLGF were maintained during treatment (Fig. 3a). VEGF concentrations in the serum also increased after initiation of motesanib; however, the magnitude of this increase was smaller than the change in PLGF, and it was transient (Fig. 3b).

Mean (±SE) fold change from baseline in PLGF (a) and VEGF (b) among patients receiving motesanib in combination with paclitaxel or docetaxel. No error bars are shown where only 1 or 2 samples were evaluable for a particular time point
Tumor response
Thirty-two patients (71 %) had measurable disease at baseline, and among the 31 assessed for tumor response, 30 experienced a decrease from baseline (Fig. 4). Overall, 18 of 32 patients (56 %) achieved a confirmed partial response, 12 (38 %) had stable disease (4 for ≥24 weeks), and 1 had progressive disease (Table 4). A higher incidence of partial responses was observed among patients who received the highest dose of motesanib: 3 of 4 in Cohort A2, 8 of 13 in Cohort B2, and 5 of 9 in Cohort B3. The clinical benefit rate (complete response + partial response + stable disease ≥ 24 weeks) was 69 %. The Kaplan–Meier estimate of the median duration of response was between 5 and 6.3 months (Table 4). Of the 13 patients with nonmeasurable disease at baseline, 10 had stable disease, 5 with stable disease ≥24 weeks, and 2 had progressive disease (Table 4).

Change from baseline in tumor measurements among patients with measurable disease at baseline. Cohorts: A1 motesanib 50 mg QD + paclitaxel 90 mg/m2; A2 motesanib 125 mg QD + paclitaxel 90 mg/m2; B1 motesanib 50 mg QD + docetaxel 100 mg/m2; B2 motesanib 125 mg QD + docetaxel 100 mg/m2; B3 motesanib 125 mg QD + docetaxel 75 mg/m2. One patient in Cohort A2 had no response assessment. SLD sum of longest diameters
Progression-free survival
At the time of this analysis, 29 patients had had PFS events (Cohort A1, n = 3; A2, n = 1; B1, n = 3, B2, n = 14, B3, n = 17). The median PFS (95 % CI) was 9.3 (3.1–21.0), 5.9 (3.4–not estimable), 6.3 (4.5–7.6), and 11.3 (5.2–12.5) months for Cohorts A1, B1, B2, and B3, respectively. Because only 1 PFS event occurred in Cohort A2 before the data cutoff, median PFS could not be evaluated for this cohort. Among the other 4 patients, 1 had a PFS event at 5.7 months, and the other 3 were on study for 5.6, 15.0, and 15.2 months without progression.
Discussion
A number of anti-VEGF pathway agents have been shown to improve outcomes for patients with MBC when used in combination with conventional chemotherapy [5]. In this phase 1b study, 45 patients with MBC received 2 different doses of motesanib in combination with either paclitaxel or docetaxel. No new safety signals were observed beyond those already demonstrated in prior phase 1 studies with single-agent motesanib. The MTD of motesanib in combination with either paclitaxel or docetaxel was 125 mg QD, consistent with that reported for single-agent motesanib in patients with advanced solid tumors [7] and in combination with platinum-containing chemotherapy and/or panitumumab in patients with NSCLC [9].
In this study, AEs were noted that have been observed in previous motesanib clinical trials: hypertension, deep vein thrombosis, and cholecystitis. Of note, hypertension, thromboembolic events, and bleeding events are known effects of VEGF(R) inhibitors [15]. In this study, 2 patients (4 %) experienced grade 3 hypertension that resulted in dose modifications. The incidence of grade 3 hypertension was less than that observed in the motesanib phase 1b NSCLC trial [9] and in a phase 3 trial of bevacizumab combined with paclitaxel in MBC [16] and the same as that observed in a phase 2 trial of bevacizumab in combination with docetaxel in MBC [17]. The overall incidence of grade ≥3 thromboembolic events (2 %) was less than observed in the motesanib phase 1b NSCLC trial and similar to that observed in the aforementioned bevacizumab studies. There were no grade ≥3 bleeding events in the current trial; grade 1 epistaxis was observed, occurring in 22 % of patients overall.
Grade 3 cholecystitis resulted in the discontinuation of treatment for 2 (4 %) patients in the study. Cholecystitis was previously reported in a phase 1b study that evaluated motesanib in NSCLC [9] and in a study with the VEGFR inhibitor sunitinib in renal cell carcinoma [18]. The etiology of this toxicity is not known, and patients should be evaluated to exclude preexisting gallbladder abnormalities before receiving motesanib and monitored for unexpected abdominal symptoms.
Paclitaxel and docetaxel had minimal effects on the pharmacokinetics of motesanib. Similarly, paclitaxel had minimal effect on motesanib pharmacokinetic parameters in patients with NSCLC [9]. In this study, paclitaxel C max and AUC0-inf values were generally higher (20–45 %) after exposure to motesanib. Similar results were observed at the 125-mg QD motesanib dose in patients with NSCLC [9]. This effect may be due to the mild inhibitory effects of motesanib on cytochrome P450 3A4 [19], which is involved in the metabolism of paclitaxel [20, 21]. Despite this effect, there appeared to be no impact on paclitaxel-related toxicities. Docetaxel AUC0-inf values were generally similar, with or without coadministration of motesanib. In contrast, docetaxel C max values were higher after exposure to motesanib in some patients likely due in part to differences in the infusion duration: the median infusion duration was slightly shorter during cycle 2 versus 1 for some patients, particularly in those in Cohort B3. Due to high interpatient variability, these results should be interpreted with caution.
The biomarker analysis showed a sustained increase in PLGF in response to motesanib treatment. These data are consistent with previous biomarker analyses of motesanib studies in various tumor types, including breast cancer [14, 22, 23]. One of those studies initially showed evidence suggesting that change in PLGF may be a predictor of response to motesanib treatment in patients with MBC [22]. However, in the large phase 3 MONET1 study of motesanib plus carboplatin/paclitaxel in patients with nonsquamous NSCLC, which prospectively assessed associations between change in PLGF and overall survival, no association between PLGF change and outcomes was identified [24].
Although the patient numbers in our study were small, the objective response rate of 56 % among patients with measurable disease is promising. To put this result into perspective, a phase 3 study of bevacizumab plus paclitaxel in previously untreated patients with MBC reported a response rate of 49.2 % among patients with measurable disease [16]. In addition, bevacizumab plus paclitaxel prolonged median PFS compared with paclitaxel alone (11.8 vs. 5.9 months) [16].
More recently, the results of 3 large phase 3 placebo-controlled trials in patients with human epidermal growth factor receptor 2-negative MBC were reported: bevacizumab or placebo plus docetaxel (AVADO) [25], bevacizumab or placebo plus anthracycline- or taxane-based chemotherapy or capecitabine (RIBBON-1) [26], and motesanib or bevacizumab or placebo plus paclitaxel (TRIO 010) [27]. The results of AVADO and RIBBON-1 showed that the addition of bevacizumab resulted in a statistically significant, although modest (1.0–2.9 months), prolongation of PFS. In TRIO 010, the addition of motesanib did not statistically significantly improve objective response rate in comparison to paclitaxel alone, although there was a trend toward superiority, favoring motesanib plus paclitaxel (49 %) and bevacizumab plus paclitaxel (52 %) compared with single-agent paclitaxel (41 %).
Despite the somewhat disappointing results of AVADO, RIBBON-1, and TRIO 010, VEGF(R) inhibitors have proven benefit in other human cancers. Further, a benefit of antiangiogenesis therapies clearly exists in breast cancer and remains an area of active investigation. Motesanib inhibits VEGFR1, VEGFR-2, and VEGFR-3 as well as PDGFR and Kit, potentially offering additional benefit not only by blocking angiogenesis but also by reducing lymphangiogenesis, and potentially tumor growth [28–31].
In summary, in the present phase 1b study, motesanib in combination with taxanes was tolerable and showed a high response rate in this population of patients with MBC. These data warrant further exploration of motesanib in the treatment of patients with breast cancer.
References
Guarneri V, Conte P (2009) Metastatic breast cancer: therapeutic options according to molecular subtypes and prior adjuvant therapy. Oncologist 14(7):645–656. doi:10.1634/theoncologist.2009-0078
Gordon MS, Mendelson DS, Kato G (2009) Tumor angiogenesis and novel antiangiogenic strategies. Int J Cancer 126(8):1777–1787. doi:10.1002/ijc.25026
Foekens JA, Peters HA, Grebenchtchikov N, Look MP, Meijer-van Gelder ME, Geurts-Moespot A, van der Kwast TH, Sweep CG, Klijn JG (2001) High tumor levels of vascular endothelial growth factor predict poor response to systemic therapy in advanced breast cancer. Cancer Res 61(14):5407–5414
Linderholm B, Grankvist K, Wilking N, Johansson M, Tavelin B, Henriksson R (2000) Correlation of vascular endothelial growth factor content with recurrences, survival, and first relapse site in primary node-positive breast carcinoma after adjuvant treatment. J Clin Oncol 18(7):1423–1431
Giovannini M, Aldrighetti D, Zucchinelli P, Belli C, Villa E (2010) Antiangiogenic strategies in breast cancer management. Crit Rev Oncol Hematol 76(1):13–35. doi:10.1016/j.critrevonc.2009.12.004
Polverino A, Coxon A, Starnes C, Diaz Z, DeMelfi T, Wang L, Bready J, Estrada J, Cattley R, Kaufman S, Chen D, Gan Y, Kumar G, Meyer J, Neervannan S, Alva G, Talvenheimo J, Montestruque S, Tasker A, Patel V, Radinsky R, Kendall R (2006) AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res 66(17):8715–8721. doi:10.1158/0008-5472.can-05-4665
Rosen LS, Kurzrock R, Mulay M, Van Vugt A, Purdom M, Ng C, Silverman J, Koutsoukos A, Sun YN, Bass MB, Xu RY, Polverino A, Wiezorek JS, Chang DD, Benjamin R, Herbst RS (2007) Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J Clin Oncol 25(17):2369–2376. doi:10.1200/JCO.2006.07.8170
Sherman SI, Wirth LJ, Droz JP, Hofmann M, Bastholt L, Martins RG, Licitra L, Eschenberg MJ, Sun YN, Juan T, Stepan DE, Schlumberger MJ (2008) Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med 359(1):31–42. doi:10.1056/NEJMoa075853
Blumenschein GR Jr, Reckamp K, Stephenson GJ, O’Rourke T, Gladish G, McGreivy J, Sun YN, Ye Y, Parson M, Sandler A (2010) Phase 1b study of motesanib, an oral angiogenesis inhibitor, in combination with carboplatin/paclitaxel and/or panitumumab for the treatment of advanced non-small cell lung cancer. Clin Cancer Res 16(1):279–290. doi:10.1158/1078-0432.ccr-09-1675
Coxon A, Bush T, Saffran D, Kaufman S, Belmontes B, Rex K, Hughes P, Caenepeel S, Rottman JB, Tasker A, Patel V, Kendall R, Radinsky R, Polverino A (2009) Broad antitumor activity in breast cancer xenografts by motesanib, a highly selective, oral inhibitor of vascular endothelial growth factor, platelet-derived growth factor, and Kit receptors. Clin Cancer Res 15(1):110–118. doi:10.1158/1078-0432.CCR-08-1155
Sweeney CJ, Miller KD, Sissons SE, Nozaki S, Heilman DK, Shen J, Sledge GW Jr (2001) The antiangiogenic property of docetaxel is synergistic with a recombinant humanized monoclonal antibody against vascular endothelial growth factor or 2-methoxyestradiol but antagonized by endothelial growth factors. Cancer Res 61(8):3369–3372
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92(3):205–216
Cancer Therapy Evaluation Program (2006) Common Terminology Criteria for Adverse Events v3.0 (CTCAE). http://ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed 10 April 2012
Bass MB, Sherman SI, Schlumberger MJ, Davis MT, Kivman L, Khoo HM, Notari KH, Peach M, Hei YJ, Patterson SD (2010) Biomarkers as predictors of response to treatment with motesanib in patients with progressive advanced thyroid cancer. J Clin Endocrinol Metab 95(11):5018–5027. doi:10.1210/jc.2010-0947
Eskens FA, Verweij J (2006) The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. Eur J Cancer 42(18):3127–3139. doi:10.1016/j.ejca.2006.09.015
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357(26):2666–2676. doi:10.1056/NEJMoa072113
Ramaswamy B, Elias AD, Kelbick NT, Dodley A, Morrow M, Hauger M, Allen J, Rhoades C, Kendra K, Chen HX, Eckhardt SG, Shapiro CL (2006) Phase II trial of bevacizumab in combination with weekly docetaxel in metastatic breast cancer patients. Clin Cancer Res 12(10):3124–3129. doi:10.1158/1078-0432.CCR-05-2603
Motzer RJ, Rini BI, Bukowski RM, Curti BD, George DJ, Hudes GR, Redman BG, Margolin KA, Merchan JR, Wilding G, Ginsberg MS, Bacik J, Kim ST, Baum CM, Michaelson MD (2006) Sunitinib in patients with metastatic renal cell carcinoma. JAMA 295(21):2516–2524. doi:10.1001/jama.295.21.2516
Yan L, Wong S, Wathen L, Chang D, Ni L, Ingram M, Parson M, Rosen L (2005) The pharmacokinetic (PK) effect of AMG 706 on CYP3A activity evaluated by use of oral midazolam as probe in patients with advanced solid tumors [abstract]. J Clin Oncol 23(Suppl 16):3178
Rochat B (2005) Role of cytochrome P450 activity in the fate of anticancer agents and in drug resistance: focus on tamoxifen, paclitaxel and imatinib metabolism. Clin Pharmacokinet 44(4):349–366
Scripture CD, Sparreboom A, Figg WD (2005) Modulation of cytochrome P450 activity: implications for cancer therapy. Lancet Oncol 6(10):780–789. doi:10.1016/S1470-2045(05)70388-0
Patterson SD, Davis MT, Mackey J, Martin M, Hei Y-J, Bass MB (2010) Biomarkers as potential predictors of response to treatment with motesanib or bevacizumab in combination with paclitaxel (P) in patients (Pts) with locally recurrent or advanced metastatic breast cancer [abstract/poster]. J Clin Oncol 28(Suppl 7):1048
Bass MB, Davis MT, Kivman L, Khoo H-M, Notari K, Blumenschein GR Jr, Mackey J, Sherman SI, Hei Y-J, Patterson SD (2010) Placental growth factor as a marker of therapeutic response to treatment with motesanib in patients with progressive advanced thyroid cancer, advanced nonsquamous non-small-cell lung cancer, and locally recurrent or advanced metastatic breast cancer [abstract]. J Clin Oncol 28(Suppl 7):3037
Scagliotti G, Vynnychenko I, Park K, Ichinose Y, Kubota K, Blackhall F, Pirker R, Galiulin R, Ciuleanu T, Sydorenko O, Dediu M, Papai-Szekely Z, Martinez Banaclocha N, McCoy S, Yao B, Hei YJ, Galimi F, Spigel DR (2012) International, randomized, placebo-controlled, double-blind phase III study of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous non-small-cell lung cancer: MONET1. J Clin Oncol. doi:10.1200/JCO.2011.41.4987
Miles DW, Chan A, Dirix LY, Cortes J, Pivot X, Tomczak P, Delozier T, Sohn JH, Provencher L, Puglisi F, Harbeck N, Steger GG, Schneeweiss A, Wardley AM, Chlistalla A, Romieu G (2010) Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol 28(20):3239–3247. doi:10.1200/JCO.2008.21.6457
Robert NJ, Dieras V, Glaspy J, Brufsky A, Bondarenko I, Lipatov O, Perez E, Yardley D, Chan Y, Zhou X, Phan S, O’Shaughnessy J (2011) RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic beast cancer. J Clin Oncol 29(10):1252–1260. doi:10.1200/JCO.2010.28.0982
Martin M, Roche H, Pinter T, Crown J, Kennedy MJ, Provencher L, Priou F, Eiermann W, Adrover E, Lang I, Ramos M, Latreille J, Jagiello-Gruszfeld A, Pienkowski T, Alba E, Snyder R, Almel S, Rolski J, Munoz M, Moroose R, Hurvitz S, Banos A, Adewoye H, Hei YJ, Lindsay MA, Rupin M, Cabaribere D, Lemmerick Y, Mackey JR, TRIO 010 investigators (2011) Motesanib, or open-label bevacizumab, in combination with paclitaxel, as first-line treatment for HER2-negative locally recurrent or metastatic breast cancer: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Oncol 12(4):369–376. doi:10.1016/S1470-2045(11)70037-7
Song S, Ewald AJ, Stallcup W, Werb Z, Bergers G (2005) PDGFRbeta+ perivascular progenitor cells in tumours regulate pericyte differentiation and vascular survival. Nat Cell Biol 7(9):870–879. doi:10.1038/ncb1288
Dvorak HF (2002) Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol 20(21):4368–4380
Heinrich MC, Blanke CD, Druker BJ, Corless CL (2002) Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J Clin Oncol 20(6):1692–1703
Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, Hung MC, Kuo ML (2006) The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell 9(3):209–223. doi:10.1016/j.ccr.2006.02.018
Acknowledgments
The authors would like to thank Bernd Bruenner (Amgen Inc.) and Cindy Kitahara (Amgen Inc.) for bioanalytical sample analysis; Michael B. Bass (Amgen Inc.) for biomarker analysis; and Kathryn Boorer (Amgen Inc.) for writing assistance. This study was funded by Amgen Inc.
Disclosure
This study was funded by Amgen Inc. REMUNERATION: Adeboye H. Adewoye (Amgen), Rebeca Melara (Amgen), and Yining Ye (Amgen), and Robert Sikorski (formerly Amgen). CONSULTANT/ADVISORY ROLE: Richard De Boer (Amgen, Sanofi-Aventis), Arlene Chan (Amgen, Sanofi-Aventis), Peter A. Kaufman (Amgen). STOCK OWNERSHIP: Adeboye H. Adewoye (Amgen), Peter A. Kaufman (Amgen), Rebeca Melara (Amgen), Yining Ye (Amgen). FUNDING: Arlene Chan (Amgen, Sanofi-Aventis), Richard De Boer (Amgen), Peter A. Kaufman (Amgen), and Shane White (Amgen).
Ethical standards
The experiments as described in this manuscript comply with the current laws of the countries in which they were performed.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
De Boer, R.H., Kotasek, D., White, S. et al. Phase 1b dose-finding study of motesanib with docetaxel or paclitaxel in patients with metastatic breast cancer. Breast Cancer Res Treat 135, 241–252 (2012). https://doi.org/10.1007/s10549-012-2135-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10549-012-2135-0