
Abstract
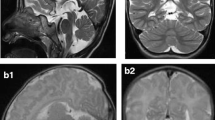
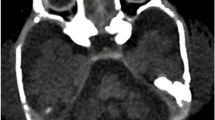
Pontocerebellar hypoplasia type 6 (PCH6) (MIM #611523) is a recently described disorder caused by mutations in RARS2 (MIM *611524), the gene encoding mitochondrial arginyl-transfer RNA (tRNA) synthetase, a protein essential for translation of all mitochondrially synthesised proteins. This case confirms that progressive cerebellar and cerebral atrophy with microcephaly and complex epilepsy are characteristic features of PCH6. Additional features of PCH subtypes 2 and 4, including severe dystonia, optic atrophy and thinning of the corpus callosum, are demonstrated. Congenital lactic acidosis can be present, but respiratory chain dysfunction may be mild or absent, suggesting that disordered mitochondrial messenger RNA (mRNA) translation may not be the only mechanism of impairment or that a secondary mechanism exists to allow some translation. We report two novel mutations and expand the phenotypic spectrum of this likely underdiagnosed PCH variant, where recognition of the characteristic neuroradiological phenotype could potentially expedite genetic diagnosis and limit invasive investigations.

Similar content being viewed by others

References
Antonellis A, Green ED (2008) The role of aminoacyl-tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 9:87–107
Antonellis A, Ellsworth RE, Sambuughin N et al (2003) Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72:1293–1299
Barkovich JA (1998) Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol 19:1389–1396
Belostotsky R, Ben-Shalom E, Rinat C et al (2011) Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA syndrome. Am J Hum Genet 88:193–200
Budde BS, Namavar Y, Barth PG et al (2008) tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet 40:1113–1118
Cavarelli J, Delagoutte B, Eriani G et al (1998) L-arginine recognition by yeast arginyl-tRNA synthetase. EMBO J 17:5438–5448
Chang BS, Piao X, Bodell A et al (2003) Bilateral frontoparietal polymicrogyria: clinical and radiological features in 10 families with linkage to chromosome 16. Ann Neurol 53:596–606
Clement E, Mercuri E, Godfrey C et al (2008) Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol 64:573–582
Delagoutte B, Moras D, Caravelli J (2000) tRNA aminoacylation by arginyl-tRNA synthetase: induced conformations during substrates binding. EMBO J 21:5599–5610
Durmaz B, Wollnik B, Cogulu O et al (2009) Pontocerebellar hypoplasia type III (CLAM): extended phenotype and novel molecular findings. J Neurol 256:416–419
Edvardson S, Shaag A, Kolesnikova O et al (2007) Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am J Hum Genet 81:857–862
Garcia-Cazorla A, Duarte S, Serrano M et al (2008) Mitochondrial diseases mimicking neurotransmitter defects. Mitochondrion 8:273–278
Götz A, Tyynismaa H, Euro L et al (2011) Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet 88:635–642
Guo M, Yang XL, Schimmel P (2010) New functions of aminoacyl tRNA synthetases. Nat Rev Mol Cell Biol 11:668–674
Hargreaves P, Rahman S, Guthrie P et al (2002) Diagnostic value of succinate ubiquinone reductase activity in the identification of patients with mitochondrial DNA depletion. J Inherit Metab Dis 25:7–16
Hausmann CD, Ibba M (2008) Aminoacyl-tRNA synthetase complexes: molecular multitasking revealed. FEMS Microbiol Rev 32:705–721
Jordanova A, Irobi J, Thomas FP et al (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Toot neuropathy. Nat Genet 38:197–202
Latour P, Thauvin-Robinet C, Baudelet-Méry C et al (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet 86:77–82
Namavar Y, Chitayat D, Barth PG et al (2011a) TSEN54 mutations cause pontocerebellar hypoplasia type 5. Eur J Hum Genet 19:724–726
Namavar Y, Barth P, Kasher P et al (2011b) Clinical, neuroradiological and genetic findings in pontocerebellar hypoplasia. Brain 134:143–156
Ng PC, Henikoff S (2003) SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31:3812–3814
Parsyan A, Svitkin Y, Shahbazian D et al (2011) mRNA helicases: the tacticians of translational control. Nat Rev Mol Cell Biol 12:235–245
Patel MS, Becker LE, Toi A et al (2006) Severe, fetal-onset form of olivopontocerebellar hypoplasia in three sibs: PCH type 5? Am J Med Genet 140A:594–603
Piao X, Chang BS, Bodell A et al (2005) Genotype-phenotype analysis of human frontoparietal polymicrogyria syndromes. Ann Neurol 8:680–687
Pierce SB, Chisholm KM, Lynch ED et al (2011) Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc Natl Acad Sci USA 108:6543–6548
Rahman S, Hanna MG (2009) Diagnosis and therapy in neuromuscular disorders: diagnosis and new treatments in mitochondrial diseases. J Neurol Neurosurg Psychiatry 80:943–953
Rajab A, Mochida GH, Hill A et al (2003) A novel form of pontocerebellar hypoplasia maps to chromosome 7q11-21. Neurology 60:1664–1667
Ramensky V, Bork P, Sunyaev S (2002) Human non-synonymous SNPs: server and survey. Nucleic Acids Res 30:3894–3900
Rankin J, Brown R, Dobyns WB et al (2010) Pontocerebellar hypoplasia type 6: a British case with PEHO-like features. Am J Med Genet A 152A:2079–2084
Renbaum P, Kellerman E, Jaron R et al (2009) Spinal muscular atrophy with pontocerebellar hypoplasia is caused by a mutation in the VRK1 gene. Am J Hum Genet 85:281–289
Riley LG, Cooper S, Hickey P et al (2010) Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia–MLASA syndrome. Am J Hum Genet 87:52–59
Scheper GC, van der Klok T, van Andel RJ et al (2007) Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat Genet 39:534–539
Stum M, McLaughlin HM, Kleinbrink EL et al (2011) An assessment of mechanisms underlying peripheral axonal degeneration caused by aminoacyl-tRNA synthetase mutations. Mol Cell Neurosci 46:432–443
Valbuena A, Sanz-García M, López-Sánchez I et al (2011) Roles of VRK1 as a new player in the control of biological processes required for cell division. Cell Signal. Apr 14 [Epub ahead of print]
Zelnik N, Dobyns WB, Forem S et al (1996) Congenital pontocerebellar atrophy in three patients: clinical, radiologic and etiologic considerations. Neuroradiology 38:684–687
Acknowledgement
We acknowledge our patient’s family for allowing us to publish her medical information and MRI images. Shamima Rahman is funded by Great Ormond Street Hospital Children’s Charity.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: John Christodoulou
Competing interest: None declared.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Figure 1
(DOC 95 kb)
Rights and permissions
About this article
Cite this article
Glamuzina, E., Brown, R., Hogarth, K. et al. Further delineation of pontocerebellar hypoplasia type 6 due to mutations in the gene encoding mitochondrial arginyl-tRNA synthetase, RARS2 . J Inherit Metab Dis 35, 459–467 (2012). https://doi.org/10.1007/s10545-011-9413-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-011-9413-6