
Abstract
Mitochondrial fatty acid oxidation represents an important pathway for energy generation during periods of increased energy demand such as fasting, febrile illness and muscular exertion. In liver, the primary end products of the pathway are ketone bodies, which are released into the circulation and provide energy to tissues that are not able to oxidize fatty acids such as brain. Other tissues, such as cardiac and skeletal muscle are capable of direct utilization of the fatty acids as sources of energy. This article provides an overview of the pathogenesis of fatty acid oxidation disorders. It describes the different tissue involvement with the disease processes and correlates disease phenotype with the nature of the genetic defect for the known disorders of the pathway.
Similar content being viewed by others
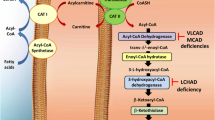
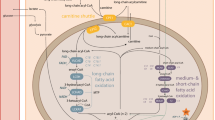

Abbreviations
- CACT:
-
carnitine-acylcarnitine translocase
- CPT:
-
carnitine palmitoyltransferase
- FAO:
-
fatty acid oxidation
- GDH:
-
glutamate dehydrogenase
- LCHAD:
-
long-chain 3-hydroxyacyl-CoA dehydrogenase
- M/SCHAD:
-
medium- and short-chain 3-hydroxyacyl-CoA dehydrogenase
- MCAD:
-
medium-chain acyl-CoA dehydrogenase
- SCAD:
-
short-chain acyl-CoA dehydrogenase
- TFP:
-
(mitochondrial) trifunctional protein
- VLCAD:
-
very long-chain acyl-CoA dehydrogenase
References
Bennett MJ, Russell LK, Tokunaga C et al (2006) Reye-like syndrome resulting from novel missense mutations in mitochondrial medium- and short-chain l-3-hydroxy acyl-CoA dehydrogenase. Mol Genet Metab 89:74–79
Brown NF, Mullur RS, Subramanian I et al (2001) Molecular characterization of L-CPT1 deficiency in six patients: insights into function of the native enzyme. J Lipid Res 42:1134–1142
Carruth A, Strauss A, Bennett M, Narayan S, Ernst LM (2009) Mutations in long-chain 3-hydroxyacyl-CoA dehydrogenase and placental maternal floor infarct/massive perivillous fibrin deposition. Proceedings of the Society for Pediatric Pathology Interim Meeting, October, 2009, Philadelphia, PA [abstract]
Clayton PT, Eaton S, Aynsley-Green A et al (2001) Hyperinsulinism in short-chain l-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of β-oxidation in insulin secretion. J Clin Invest 108:457–465
den Boer MEJ, Wanders RJA, Morris AAM, IJlst L, Heymans HS, Wijburg FA (2002) LCHAD deficiency: clinical presentation and follow up in 50 patients. Pediatrics 109:99–104
Filling C, Keller B, Hirschberg D et al (2008) Role of short-chain hydroxyacyl-CoA dehydrogenase in SCHAD deficiency. Biochem Biophys Res Commun 368:6–11
Gillingham MB, Banta-Wright SA, Hermerath CA et al (2006) CPT1A P479L variant identified in Alaska natives by expanded newborn screening. J Inherit Metab Dis 29(Suppl 1; 85) [abstract]
Greenberg CR, Dilling LA, Thompson GR et al (2009) The paradox of the carnitine palmitoyltransferase type 1a P479L variant in Canadian Aboriginal populations. Mol Genet Metab 96:201–207
Hussain K, Clayton PT, Krywawych S et al (2005) Hyperinsulinism of infancy associated with a novel splice site mutation in the SCHAD gene. J Pediatr 146:706–708
Iafolla AK, Thompson RJ, Roe CR (1994) Medium-chain acyl-CoA dehydrogenase deficiency: clinical course in 120 affected children. J Pediatr 124:409–415
Ibdah J, Bennett MJ, Rinaldo P et al (1999) A fetal fatty acid oxidation disorder as a cause of liver disease in pregnant women. N Engl J Med 340:1723–1731
Jethva R, Ficicioglu C (2008) Clinical outcomes of infants with short-chain acyl-coenzyme A dehydrogenase deficiency (SCADD) detected by newborn screening. Mol Genet Metab 95:241–242
Jethva R, Bennett MJ, Vockley J (2008) Short-chain acyl-coenzyme A dehydrogenase deficiency. Mol Genet Metab 95:195–200
Kapoor RR, James C, Flanagan SE, Ellard S, Eaton S, Hussain K (2009) 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinaemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. J Clin Endocrinol Metab 94:2221–2225
Matern D, Shehata BM, Shekhawat P, Strauss AW, Bennett MJ, Rinaldo P (2001) Placental floor infarction complicating the pregnancy of a fetus with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Mol Genet Metab 72:265–268
McGarry JD, Brown NF (1997) The mitochondrial carnitine palmitoyltransferase system: from concept to molecular analysis. Eur J Biochem 244:1–6
Molven A, Matre GE, Duran M et al (2004) Familial hyperinsulinemic hypoglycemia caused by a defect in the SCHAD enzyme of mitochondrial fatty acid oxidation. Diabetes 53:221–227
Oey NA, Ruiter JPN, Attie-Bitach T, IJlst L, Wanders RJA, Wijburg FA (2006) Fatty acid oxidation in the human fetus: implications for fetal and adult disease. J Inherit Metab Dis 29:71–75
Olpin SE, Afifi A, Clark S et al (2003) Mutation and biochemical analysis in carnitine palmitoyltransferase type II (CPT II) deficiency. J Inherit Metab Dis 26:543–557
Pedersen CB, Kolvraa S, Kolvraa A et al (2008) The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum Genet 124:43–56
Price NT, van der Leij FR, Jackson VN et al (2002) Anovel brain-expressed protein related to carnitine palmitoyltransferase 1. Genomics 80:433–442
Rinaldo P, Matern D, Bennett MJ (2002) Fatty acid oxidation disorders. Annu Rev Physiol 64:477–502
Shekhawat P, Bennett MJ, Sadovsky V, Nelson DM, Rakheja D, Strauss AW (2003) Human placenta metabolizes fatty acids: implications for fetal fatty acid oxidation disorders and maternal liver diseases. Am J Physiol Endocrinol Metabol 284:E1098–E1105
Shekhawat P, Sonne S, Matern D et al (2007) Spontaneous development of intestinal and colonic atrophy and inflammation in the carnitine deficient jvs (OCT2N−/−) mice. Mol Genet Metab 92:315–324
Snider MD, McGarry JD, Hanson RW (2006) Lipid metabolism 1: synthesis, storage and utilization of fatty acids and triacylglycerols. In: Devlin TM (ed) Textbook of biochemistry with clinical correlations, 6th edn. Wiley, Hoboken, pp 662–694
Stanley CA, Lieu YK, Hsu BY et al (1998) Hyperinsulinism and hyperammonemia in infants with regulatory mutations of the glutamate dehydrogenase gene. N Engl J Med 338:1352–1357
Strauss AW, Andresen BS, Bennett MJ (2009) Mitochondrial fatty acid oxidation defects. In: Sarafoglou K, Hoffmann GF, Roth KS (eds) Pediatric endocrinology and inborn errors of metabolism. McGraw-Hill, New York, pp 51–70
Thuillier L, Rostane H, Droin V et al (2003) Correlation between genotype, metabolic data, and clinical presentation in carnitine palmitoyltransferase 2 deficiency. Hum Mutat 21:493–501
Tyni T, Kivela T, Lappi M, Summainen P, Nikoskelainen E, Pihko H (1998) Ophthalmic findings in LCHAD deficiency caused by the G1528C mutation. Ophthalmology 105:810–824
Wilcken B, Leung KC, Hammond J, Kamath R, Leonard JV (1993) Pregnancy and fetal LCHAD deficiency. Lancet 341:407–408
Wilcken B, Haas M, Joy P et al (2007) Outcome of neonatal screening for medium-chain acyl-CoA dehydrogenase deficiency in Australia: a cohort study. Lancet 369:37–42
Wood PA, Amendt BA, Rhead WJ, Millington DS, Inoue F, Armstrong D (1989) Short-chain acyl-coenzyme A deficiency in mice. Pediatr Res 25:38–43
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by: Ertan Mayatepek
Competing interest: None declared.
Rights and permissions
About this article
Cite this article
Bennett, M.J. Pathophysiology of fatty acid oxidation disorders. J Inherit Metab Dis 33, 533–537 (2010). https://doi.org/10.1007/s10545-010-9170-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-010-9170-y