
Summary
In view of the major improvements in treatment, it has become increasingly important that in order for first-line physicians not to miss a treatable disorder they should be able initiate a simple method of clinical screening, particularly in the emergency room. We present a simplified classification of treatable inborn errors of metabolism in three groups. Group 1 includes inborn errors of intermediary metabolism that give rise to an acute or chronic intoxication. It encompasses aminoacidopathies, organic acidurias, urea cycle disorders, sugar intolerances, metal disorders and porphyrias. Clinical expression can be acute or systemic or can involve a specific organ, and can strike in the neonatal period or later and intermittently from infancy to late adulthood. Most of these disorders are treatable and require the emergency removal of the toxin by special diets, extracorporeal procedures, cleansing drugs or vitamins. Group 2 includes inborn errors of intermediary metabolism that affect the cytoplasmic and mitochondrial energetic processes. Cytoplasmic defects encompass those affecting glycolysis, glycogenosis, gluconeogenesis, hyperinsulinisms, and creatine and pentose phosphate pathways; the latter are untreatable. Mitochondrial defects include respiratory chain disorders, and Krebs cycle and pyruvate oxidation defects, mostly untreatable, and disorders of fatty acid oxidation and ketone bodies that are treatable. Group 3 involves cellular organelles and includes lysosomal, peroxisomal, glycosylation, and cholesterol synthesis defects. Among these, some lysosomal disorders can be efficiently treated by enzyme replacement or substrate reduction therapies. Physicians can be faced with the possibility of a treatable inborn error in an emergency, either in the neonatal period or late in infancy to adulthood, or as chronic and progressive symptoms – general (failure to thrive), neurological, or specific for various organs or systems. These symptoms are summarized in four tables. In addition, an extensive list of medications used in the treatment of inborn errors is presented.
Similar content being viewed by others
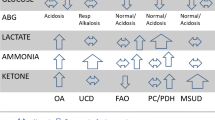


Abbreviations
- 3PGD:
-
3-phosphoglycerate dehydrogenase
- BCAA:
-
branched-chain amino acid
- BRBGD:
-
biotin-responsive basal ganglia disease
- Cbl:
-
cobalamin
- CDG:
-
congenital disorder of glycosylation
- CPT I:
-
carnitine palmitoyltransferase type I
- CPT II:
-
carnitine palmitoyltransferase type II
- CTX:
-
cerebrotendinous xanthomatosis
- FAO:
-
fatty acid oxidation
- GTP:
-
guanosine triphosphate
- HELLP:
-
haemolysis, elevated liver function, low platelets
- HFI:
-
hereditary fructose intolerance
- IE:
-
inborn error
- IEM:
-
inborn error of metabolism
- LPI:
-
lysinuric protein intolerance
- HMG CoA:
-
3-hydroxy-3-methylglutaryl coenzyme A
- IVA:
-
isovaleric acidaemia
- LCHADD:
-
long-chain 3-hydroxy-acyl-CoA dehydrogenase deficiency
- MCD:
-
multiple carboxylase deficiency
- MMA:
-
methylmalonic acidaemia
- MSUD:
-
maple syrup urine disease
- MTHFR:
-
methylene tetrahydrofolate reductase
- OA:
-
organic aciduria
- OTC:
-
ornithine transcarbamylase
- PA:
-
propionic acidaemia
- PC:
-
pyruvate carboxylase
- PDH:
-
pyruvate dehydrogenase
- PKU:
-
phenylketonuria
- PNPO:
-
pyridox(am)ine-5'-phosphate oxidase
- PTP:
-
6-pyruvoyltetrahydropterin synthase
- TFP:
-
trifunctional protein
- TH:
-
tyrosine hydroxylase
- TL:
-
carnitine acyltranslocase
- UCD:
-
urea cycle disorders
- VLCADD:
-
very long-chain acyl-CoA dehydrogenase deficiency
References
de Koning T (2006) Treatment with amino acids in serine deficiency disorders. J Inherit Metab Dis 29: 347–351.
Junien C (2006) Impact of diets and nutrients/drugs on early epigenetic programming. J Inherit Metab Dis 29: 359–365.
Kjaergaard S, Kristiansson B, Stibler H, et al (1998) Failure of short-term mannose therapy of patients with carbohydrate- deficient glycoprotein syndrome type 1A. Acta Paediatr 87: 884–888.
Koga Y, Akita Y, Nishioka J, et al (2005) l-Arginine improves the symptoms of strokelike episodes in MELAS. Neurology 64: 710–712.
Mayatepek E, Schroder M, Kohlmuller D, et al (1997) Continuous mannose infusion in carbohydrate-deficient glycoprotein syndrome type I. Acta Paediatr 86: 1138–1140.
Ostman-Smith I, Brown G, Johnson A, et al (1994) Dilated cardiomyopathy due to type II X-linked 3-methylglutaconic aciduria: successful treatment with pantothenic acid. Br Heart J 72: 349–353.
Quinzii C, Naini A, Salviati L, et al (2006) A mutation in para-hydroxybenzoate-polyprenyl transferase (COQ2) causes primary Coenzyme Q10 deficiency. Am J Hum Genet 78: 345–349.
Raine DN (ed.) (1975) The Treatment of Inherited Metabolic Disease. Lancaster: Medical and Technical Publishing.
Roe CR, Mochel F (2006) Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis 29: 332–340.
Saudubray JM, Desguerre I, Sedel F, Charpentier C (2006) A clinical approach to inherited metabolic disorders. In: Fernandes J, Saudubray JM, van den Berghe G, Walter J, eds. Inborn Metabolic Diseases: Diagnosis and Treatment, 4th edn. Berlin: Springer-Verlag, 2006 in press.
Valayannopoulos V, Verhoeven NV, Mention K, et al (2006) Transaldolase deficiency: a new cause of hydrops foetalis and neonatal multiorgan disease. J Pediatr, in press.
Van Spronsen FJ, Smit GPA, Erwich JJHM (2005) Inherited metabolic diseases and pregnancy. BJOG Int J Obstet Gynaecol 112: 2–11.
Walter JH, Wraith J (2006) Treatment: present status and new trends. In: Fernandes J, Saudubray JM, van den Berghe G, Walter J, eds. Inborn Metabolic Diseases: Diagnosis and Treatment. Berlin: Springer-Verlag, 4th edition, in press.
Zeng WQ, Al Yamani E, Acierno JS Jr, et al (2005) Biotin-responsive basal ganglia disease maps to 2q36.3 and is due to mutations in SLC19A3. Am J Hum Genet 77: 16–26.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Jean-Marie Saudubray
Competing interests: None declared
Rights and permissions
About this article
Cite this article
Saudubray, JM., Sedel, F. & Walter, J.H. Clinical approach to treatable inborn metabolic diseases: An introduction. J Inherit Metab Dis 29, 261–274 (2006). https://doi.org/10.1007/s10545-006-0358-0
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1007/s10545-006-0358-0