
Abstract
Alkaline-earth metal complexes of the monoanionic form of the polyether ionophore monensin A were isolated for the first time in solid state and were structurally characterized using various spectroscopic methods (IR, NMR, FAB-MS). The stoichiometric reaction of monensic acid (MonH) with M2+ (M = Mg, Ca) in the presence of an organic base leads to the formation of mononuclear complexes of composition [M(Mon)2(H2O)2]. The structures of magnesium (1) and calcium (2) monensin complexes in the solid state were established by single crystal X-ray crystallography. The complexes crystallize as [Mg(Mon)2(H2O)2]·5MeCN (1) and [Ca(Mon)2(H2O)2]·H2O·5MeCN (2) in the monoclinic P21 space group. The alkaline-earth metal ion is placed in a distorted octahedral environment, defined by two monensin anions acting as bidentate ligands in the equatorial plane of the complex as well as by two water molecules occupying the axial positions of the inner coordination sphere. The bactericidal activity of 1 and 2 was evaluated against aerobic Gram-positive microorganisms applying the double layer agar hole diffusion method.
Similar content being viewed by others

Introduction
Recently the most powerful drugs in veterinary medicine effective against coccidiosis and infections induced by Gram-positive organisms represent the group of naturally occurring polyether ionophorous antibiotics (Agtarap et al. 1967; Stern 1977; Berg and Hamill 1978; Liu et al. 1978; Chappe 1979; Long and Jeffers 1982; Westley et al. 1983; Augustine et al. 1987; Koinarski and Sherkov 1987; Folz et al. 1988; Augustine et al. 1992; Varga and Sreter 1996; Wang et al. 2006; Kevin et al. 2009). Their main representative—monensin—is well known and its ability of binding monovalent metal ions is extensively studied. The neutral ionophore complexes formed are able to penetrate the cell membrane of the microorganisms and parasites and to disturb a series of homeostatic processes after dissociation in the intracellular space causing a death of the corresponding target (Riddell 2002). Due to the high affinity of monensin to complex alkaline metal ions, it is known for a very long time to be a monovalent polyether ionophore (Lutz et al. 1971; Briggs and Hinton 1978; Duax et al. 1980; Garcla-Rosas et al. 1983; Cox et al. 1984; Walba et al. 1986; Pangborn et al. 1987; Rzeszotarska et al. 1994; Wagner-Czauderna et al. 1997; Paz et al. 2003; Huczynski et al. 2007a, b, c, 2008a, b, c, d; Yildirim et al. 2007). From the 1990s up to the present several research groups have drawn attention to the possible complexation of monensin with divalent metal ions (Hebrant et al. 1992; Mimouni et al. 1994; Dassie and Baruzzi 2002; Hamidinia et al. 2002; Huczynski et al. 2006a, b, c, d, 2007a, b, c, 2008a, b, c, d). Although there was only indirect evidence concerning the unusual coordination behaviour of monensin, it was suggested that the complexation chemistry of the ligand (and of other representatives of the monovalent polyether ionophores group) is much broader than we were previously acquainted with (Stiles et al. 1991; Hamidinia et al. 2002, 2004). The hypothesis that monensin possesses previously unknown reactivity prompted our group to start an extensive study on the coordination ability of the ligand towards di- and three-valent metal ions. In our previous publications we demonstrated that monensin could form two types of divalent transition metal complexes depending on the chemical form used in the reactions (Dorkov et al. 2008; Pantcheva et al. 2008, 2009). The compounds obtained possess enhanced antimicrobial activity towards Gram(+)-bacteria that is most likely due to the unusual coordination mode of the ligand.
In order to confirm the ability of monensin to bind divalent metal ions, we have now studied its behaviour towards alkaline-earth metals, in particularly magnesium and calcium ions. In the present paper we describe the structure elucidation of Mg2+ and Ca2+ complexes of the monoanionic form of monensic acid (MonH, Scheme 1) in the solid state by single crystal X-ray crystallography, report on their spectroscopic characterization and on the evaluation of their bactericidal properties against aerobic Gram-positive microorganisms.

The chemical structure and numbering sequence of monensic acid
Materials and methods
Chemicals
All chemicals were of reagent grade and were used as received. The commercially available sodium monensin was obtained from BIOVET, Ltd. (Bulgaria). Solvents (MeCN, MeOH, DMSO), organic base (Et4NOH) and metal salts (MgCl2·6H2O, CaCl2) were purchased from Merck.
Synthesis of monensic acid
The acidic form of monensin A monohydrate (monensic acid, MonH·H2O) was prepared from sodium monensin as previously reported (Gertenbach and Popov 1975). IR: νCOOH = 1700 cm−1. 1H-NMR (600 MHz, δ (multiplicity, intensity, J-coupling(s), assignment), CDCl3): 6.25 (br, 1-OH, 10-OH (sharp), 11-OH, H2O), 4.50 (br d, 1H, 7.9, 5-OH), 4.33 (ddd, 1H, 10.8, 5.9, 2.9, 20CH), 4.07 (dd, 1H, 11.6, 2.1, 5CH), 4.03 (d, 1H, 4.0, 17CH), 3.94 (dd, 1H, 10.5, 2.7, 21CH), 3.86 (br s, 1H, 7CH), 3.69 (d, 1H, 11.2, 26CH2′), 3.50 (d, 1H, 11.2, 26CH2″), 3.44 (dd, 1H, 10.7, 4.6, 13CH), 3.37 (s, 3H, 28OCH3), 3.22 (dd, 1H, 10.2, 2.1, 3CH), 2.62 (dq, 1H, 10.2, 6.7, 2CH), 2.25–2.13 (4H, 18CH, 15CH2′, 4CH, 19CH2′), 2.10 (m, 1H, 6CH), 1.98 (dd, 1H, 14.2, 3.5, 8CH2′), 1.97 (dd, 1H, 12.3, 8.4, 10CH2′), 1.89 (dt, 1H, 11.9, 8.7, 11CH2′), 1.74–1.52 (m, 5H, 8CH2″, 11CH2″, 10CH2″, 14CH2), 1.55 (m, 2H, 32CH2), 1.52–1.33 (m, 5H, 23CH2′, 24CH, 15CH2″, 19CH2″, 23CH2″), 1.48 (s, 3H, 31CH3), 1.35 (m, 1H, 22CH), 1.27 (d, 3H, 6.7, 27CH3), 1.10 (d, 3H, 6.9, 29CH3), 0.95 (t, 3H, 7.4, 33CH3), 0.92 (d, 3H, 6.9, 34CH3), 0.88 (d, 3H, 7.1, 30CH3), 0.87 (d, 3H, 6.4, 36CH3), 0.85 (d, 3H, 6.5, 35CH3).
Synthesis of magnesium complex, [Mg(Mon)2(H2O)2], 1
The consecutive addition of Et4NOH (0.5 mmol, 180 μl, 40% in H2O) and of freshly prepared solution of MgCl2.6H2O (0.5 mmol, 100 mg in 5 ml MeCN/MeOH, 10:1) to a solution of MonH (0.5 mmol, 336 mg in 20 ml MeCN/MeOH, 10:1) afforded the precipitation of white solids with a composition of [Mg(Mon)2(H2O)2], 1, which are insoluble in MeCN (228 mg, 65% yield). The complex was filtered off, washed with MeCN and dried over P2O5 at room temperature. Anal. Calcd for MgC72H126O24 (MW = 1400.08): H, 9.07, C, 61.77, Mg, 1.74; Found: H, 8.76, C, 60.17, Mg, 2.14%. Slow concentration of diluted reaction mixture affords the formation of colourless crystals of composition [Mg(Mon)2(H2O)2]·5MeCN. IR: νCOO asym = 1550 cm−1, νCOO sym = 1400 cm−1. 1H-NMR (600 MHz, δ (assignment), CDCl3): 10.98 (11-OH), 6.60 (10-OH), 4.98 (H2O), 4.85 (5-OH), 4.30 (20CH), 4.09 (21CH), 4.04 (17CH, 5CH), 3.73 (7CH), 3.78 (26CH2′), 3.68 (26CH2″), 3.52 (13CH), 3.33 (28OCH3), 3.24 (3CH), 2.46 (2CH), 2.33 (6CH), 2.23 (18CH), 2.17-2.04 (15CH2′, 19CH2′, 4CH), 2.00-1.94 (8CH2′, 10CH2′), 1.85 (11CH2′), 1.75–1.68 (14CH2′), 1.70–1.50 (8CH2″, 11CH2″, 10CH2″, 14CH2″, 32CH2′, 23CH2′), 1.54 (31CH3), 1.50–1.40 (19CH2″, 32CH2″, 15CH2″), 1.37 (24CH), 1.29 (23CH2″, 22CH), 1.18 (27CH3), 1.08 (29CH3), 0.98–0.82 (33CH3, 34CH3, 30CH3, 36CH3, 35CH3).
Synthesis of calcium complex, [Ca(Mon)2(H2O)2], 2
To a reaction mixture containing MonH (0.5 mmol, 336 mg in 15 ml MeCN/MeOH (10:1)) and Et4NOH (0.5 mmol, 180 μl, 40% in H2O) the addition of CaCl2 (0.5 mmol, 56 mg in 5 ml MeCN/MeOH, 10:1) led to the formation of colourless solution that slowly precipitates within 24 h (240 mg, 68% yield). The solid was filtered off, washed with MeCN and dried over P2O5 at room temperature. The elemental analysis data are in agreement with the general composition of the complex, [Ca(Mon)2(H2O)2], 2. Anal. Calcd. for CaC72H126O24 (MW = 1415.86): H, 8.97, C, 61.08, Ca, 2.83; Found: H, 8.56, C, 58.12, Ca, 3.20%. Complex 2 is soluble in MeOH, CHCl3 and octanol, and possesses very low solubility in MeCN and DMSO. IR: νCOO asym = 1560 cm−1, νCOO sym = 1400 cm−1. 1H-NMR (600 MHz, δ (assignment), CDCl3): 4.95 (5-OH), 4.32 (20CH), 4.23 (17CH), 4.04 (5CH, 21CH), 3.86–3.68 (7CH, 26CH2′), 3.56 (26CH2″), 3.50 (13CH), 3.37 (28OCH3), 3.24 (3CH), 2.48 (2CH), 2.37 (6CH), 2.30 (18CH), 2.22–2.11 (15CH2′, 19CH2′), 2.09 (4CH), 2.00–1.88 (8CH2′, 10CH2′, 11CH2′), 1.76–1.58 (8CH2″, 11CH2″, 10CH2″, 14CH2), 1.58–1.40 (32CH2, 23CH2′, 24CH, 31CH3, 15CH2″, 19CH2″), 1.40–1.26 (23CH2″, 22CH), 1.19 (27CH3), 1.12 (29CH3), 0.98–0.83 (33CH3, 34CH3, 30CH3, 36CH3, 35CH3).
Slow concentration of diluted reaction mixture (2–3 days at ambient temperature) affords the precipitation of 2 as colorless crystals. The X-ray analysis showed that 2 crystallizes as [Ca(Mon)2(H2O)2]·H2O·5MeCN.
Physical measurements
Infrared spectra (4000–400 cm−1) were recorded on a Specord 75-IR (Carl-Zeiss, Germany) in a nujol mull. FAB-MS spectra were performed using Fisons VG Autospec (Micromass Instruments, UK).
1H (600.13 MHz) and 13C (150.92 MHz) spectra were acquired on an AVANCE AV600 II+ NMR spectrometer (Bruker, Germany). All spectra were recorded in CDCl3 at room temperature. TMS was used as an internal standard for the 1H and 13C spectra. Unambiguous assignment of the signals was made on the basis of the gradient enhanced versions of COSY, TOCSY, HSQC, HMBC and ROESY experiments (Bruker pulse library programs: cosygpmfqf, dipsi2etgpsi, hsqcedetgpsisp2.2, hmbcgplpndqf, roesyph.2, 2007). The chemical shift values of the individual protons in the compounds have been determined from the HSQC spectra.
Elemental analysis data (C, H, O) were obtained with a VarioEL V5.18.0 Elemental Analyzer (Elementar Analysen Systeme GmbH, Germany). The metal content of complexes was determined by AAS on a Perkin Elmer 1100 B (Waltham, USA) after decomposition of samples with conc. HNO3 and using a stock standard solution (Merck, 1000 μg/ml); the working reference solutions were prepared after suitable dilution.
X-ray crystallography
Details concerning data collection, structure solution and refinement of complexes 1 and 2 are given in Table 1. X-ray diffraction measurements were performed on an Oxford Diffraction Xcalibur 2 diffractometer at 112 K, operating with Mo-Kα (λ = 0.71073 Å) radiation and equipped with a graphite monochromator. The structures were solved by direct methods and were refined by full-matrix least-square procedures on F 2 (Sheldrick 1990; Sheldrick 1997). All non-H atoms were refined isotropically with a riding model.
Cytotoxicity assay (determination of MIC)
Three aerobic Gram-positive microorganisms were used as test strains to evaluate the cytotoxic properties of the alkaline-earth complexes, MgCl2.6H2O and CaCl2. The bacteria Bacilus subtillis (ATCC 6633), Bacilus mycoides spp. and Sarcina lutea FDA strain PCI 1000 (ATCC 10054) were purchased from the National Bank for Industrial Microorganisms and Cell Cultures (Bulgaria). The activity of compounds is determined as their minimum inhibitory concentration [MIC, (μM)], which is the lowest concentration causing the visible inhibition of the bacteria growth. Details concerning the experimental procedures using the double layer agar hole diffusion method were carried in accordance with the literature (Andrews 2001) and were similar to those reported previously (Dorkov et al. 2008; Pantcheva et al. 2008, 2009).
Results and discussion
The reaction of monensic acid with MgCl2 or CaCl2 in the presence of an organic base leads to the formation of unique alkaline-earth metal complexes of composition [M(Mon)2(H2O)2] (M = Mg (1), Ca (2)). The reaction proceeds in a mixed solvent system (MeCN/MeOH) and at metal-to-ligand-to-base molar ratio of 1:1:1. The addition of Et4NOH is essential for the coordination of the ligand to the divalent metal ions since it facilitates deprotonation of the carboxylic function of monensic acid during the complexation. In the absence of organic base the reaction does not take place, and the use of inorganic bases such as NaOH or KOH exclusively forms the monovalent metal complexes of monensin, MonNa or MonK, respectively.
Crystal structures of [Mg(Mon)2(H2O)2], 1 and [Ca(Mon)2(H2O)2], 2
Generally, the magnesium and calcium complexes of monoanionic form of monensic acid consist of a discrete electrically neutral unit [M(Mon)2(H2O)2]. Both compounds crystalize in the monoclinic space group P21. The crystals analyzed by X-ray crystallography additionally contain solvents molecules (MeCN and/or H2O), which do not affect the coordination mode of the ligand and do not participate in the formation of intramolecular H-bonds with the main unit. Such a solvent insertion was already observed in the cases of Mn2+/Co2+ complexes of deprotonated monensic acid (Pantcheva et al. 2008) and of Ba2+ complex of lasalocid (Johnson et al. 1970a, b).
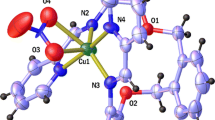
In compounds 1 and 2, two monensic anions (Mon) act as bidentate ligands occupying four places in the inner coordination sphere of the alkaline-earth metal ion. Two of the positions in the equatorial plane of the complexes are filled by deprotonated monodentate carboxylic functions of two monensic anions (COO–M bonds), and other two by methoxy groups of Mon located at the opposite end of the ligand molecule forming two dative HO → M bonds, respectively. The folding of the ligand due to its bidentate coordination mode affords its pseudo-cyclization referred to as “head-to-tail” cyclization, observed even for the free ligand in the absence of metal ions (Lutz et al. 1971). Similarly to the transition metal complexes of Mon (Pantcheva et al. 2008) and barium complex of lasalocid (Johnson et al. 1970a, b), two water molecules are also included in the structures of 1 and 2, linked by dative H2O → M bonds in axial positions with respect to the divalent metal ion and completing the octahedral environment of Mg2+/Ca2+ (Fig. 1). The data confirm that water molecules in the new monensin complexes play a dual role: first, they occupy the inner coordination sphere of the metal ion, and in the second place they stabilize the strongly folded ligand by various intramolecular H-bonds. Selected bond distances and bond angles of 1 and 2 are presented in Table 2. Intramolecular H-bonds observed are shown in Fig. 2 and Table 3, respectively. No intermolecular H-bonds were detected in the structures. The crystal packing of complexes is depicted in Fig. 3.

ORTEP of [Mg(Mon)2(H2O)2] (1) and [Ca(Mon)2(H2O)2] (2) at the 30% probability level (protons and solvent molecules are omitted for clarity)

Intramolecular bonds (in dotted line) observed in the structure of alkaline-earth complexes on the basis of complex 1 (the coordination of Mg2+ with one of the ligands is shown)

Crystal packing of complexes 1 (a) and 2 (b)
The comparison of crystallographic data for the Mn2+/Co2+ (Pantcheva et al. 2008) and Ca2+/Mg2+ complexes of monoanionic monensic acid shows that the divalent metal center reacts in a similar manner in each case both with monensin ligands and with water molecules. The M–O bond lengths decrease in the order of Ca2+ > Mn2+ > Co2+ > Mg2+ in accordance with the decrease of the metal ionic radii. The M–O bond angles of metal ion with equatorial monensin ligands forming the xy-plane of complexes deviate from the regular ones (180°) by ca. 10° and no significant difference is observed between them in dependence on the divalent metal center. In contrast, the metal-aqueous oxygen bond angles increase with decreasing metal ionic radii as follows: Ca2+ (151.2°) < Mn2+ (154.4°) < Co2+ (159.1°) < Mg2+ (160.4°). From the data collected up to now it can be concluded that the nature of the divalent metal ion (i.e. its ionic radius) does not affect significantly the equatorial planarity of complexes, but influences the bonding with axial ligands thus leading to different degrees of deformation of the octahedral environment around the divalent cationic center.
The structures of 1 and 2 have clearly been established in the solid state by single crystal X-ray diffraction, nevertheless questions concerning the coordination mode of deprotonated monensic acid especially towards Ca2+ still remain. These queries arise since calcium and sodium ions are hard acids and their ionic radii are equivalent at coordination number of 6 [1.02 Å (Na+) vs. 1.00 Å (Ca2+)], but at the same time Ca2+ does not “enter” the hydrophilic cavity of ligand at the selected reaction conditions. Although some authors suggest the formation of shell-like alkaline-earth complexes with monovalent ionophores of composition metal-to-ligand ratio = 1:1 (Huczynski et al. 2007a, b, c, 2008a, b, c, d) their isolation as solid phases was not observed in the present research. Despite these hypothesis, the magnesium (1) and calcium (2) complexes of monoanionic monensic acid of composition of M:Mon = 1:2 clearly represent the first example of alkaline-earth monensin complexes to be isolated and fully characterized in solid state.
IR spectral properties of complexes 1 and 2
The IR spectra of 1 and 2 obtained in a nujol mull are very similar to those observed for the transition metal complexes of Mon (Pantcheva et al. 2008) and the results are in agreement with the crystallographic data of new compounds. The main evidence that complexation takes place is found comparing the positions of stretching vibrations of the carboxyl function of MonH and of the carboxylate group of 1 and 2, respectively. The peak due to νCOOH of MonH (1700 cm−1) disappears in the IR spectra of 1 and 2 thus demonstrating that deprotonation of carboxyl moiety occurs during the complexation. This fact was also confirmed by the appearance of two new peaks in the IR spectra of 1 and 2, assigned to the stretching vibrations of the deprotonated carboxylic function, absorbing at 1550 cm−1 (ν asymCOO ) and 1400 cm−1 (ν symCOO ), respectively. The IR spectral data confirming the coordination both of monensin hydroxyl groups and of water ligands to Mg2+ and Ca2+ are also in accordance with the data previously reported (Pantcheva et al. 2008). The crystallization water and hydroxyl groups of monensic acid absorb at 3520 and 3320 cm−1, respectively, while in the IR spectra of 1 and 2 the next bands were found and were assigned as follows: νOHwater (3490 cm−1) and νOHOH-groups [3360, 3280 cm−1 (1); 3280 cm−1 (2)].
NMR spectra of MonH, 1 and 2
To the best of our knowledge, several research teams solved 1H- and 13C{1H}-NMR spectra of the monovalent metal complexes of Mon (Ajaz et al. 1987; Mimouni et al. 1996; Martinek et al. 2000) or its ester derivatives and their alkali complexes (Huczynski et al. 2006a, b, c, d, 2007a, b, c, 2008a, b, c, d), respectively. The presented study for a first time is dealing in details with the NMR spectra of monensic acid and its diamagnetic divalent metal derivatives. The 1H-NMR data of MonH, 1 and 2 are presented in “Materials and methods” section. 13C{1H}-NMR resonances are summarized in Table 4, following the numbering sequence of the ligand, shown in Scheme 1.
Due to the low solubility of metal complexes in MeCN, the NMR spectra were recorded in CDCl3 where OH-exchange is taking place thus significantly affecting the position of OH-groups. The 1-OH (–COOH), 10-OH and 11-OH resonances in monensic acid appear as a broad signal at 6.25 ppm and 5-OH is observed at 4.50 ppm. The major evidence that the deprotonation of carboxylic function of MonH occurs during the complexation is the disappearance of 1-OH resonance in 1H-NMR spectra of 1 and 2. At the same time a significant positive deviation in the position of 5-OH is observed whose resonance shifts from 4.50 ppm (MonH) to 4.85 ppm (1) and 4.95 ppm (2), respectively. The results obtained suggest that in solution this group is more strongly engaged in intramolecular H-bonding in the complexes than in the free acid and this was confirmed by the solid state structure of metal(II) compounds. As can be seen from the crystallographic data (Table 3; Fig. 3), 5-OH participates in three H-bonds by interacting with the aqua ligand (H2O-12), the ether oxygen O6 and the hydroxyl group 10-OH.
The main point of emphasis when discussing the 13C{1H}-NMR spectra of MonH, 1 and 2 is focused on the positions of those carbon atoms, that are closely attached to the coordination sites of the ligand and whose resonances deviate significantly in the spectra on moving from MonH to complexes 1 and 2. As can be seen (Table 4), the position of 1C shows a very significant downfield shift in the spectra of 1 and 2 comparing to MonH (Δ = 4.85 ppm (1), Δ = 4.72 ppm (2), Δ = δcomplex − δMonH). Such behaviour is in agreement with the presence of monensin deprotonated carboxylic function and reveals that both complexes retain their coordination in solution. A downfield shift in the spectra of 1 and 2 is also observed for the carbons 2, 3, 25, 27 and 29 (Δ = 0.7−2.5 ppm), which are in close vicinity of atoms O1 and O11 directly coordinated to the divalent metal ions. The formation of H-bonds between the water ligands (H2O-12 and H2O-13) and ether oxygens O5–O8 also shifts some of the carbon-13 resonances of 1 and 2 significantly. The experimental data obtained show that positive as well as negative shifts are observed depending on the extent of deformation, that the ligand endures through the complexation. The NMR data obtained for Mg- and Ca-complexes of Mon correspond closely and confirm that (i) the coordination mode of monensin is analogous in 1 and 2; (ii) the metal ions retain their linkage with bidentate monensic anions both in solid state and in solution.
FAB-MS spectra of complexes 1 and 2
The major species found in the FAB-MS spectra of complexes 1 and 2 are presented in Scheme 2. The spectra consist of a great number of signals some of which are assigned to the molecular ions of complexes after loss of water ligands and association with sodium ions, [M(C36H61O11)Na]+ (1a: M = Mg; 2a: M = Ca), as well as to the ions generated from association/dissociation processes occurring with the complex species in the gas phase (1b, 2c–f). It should be mentioned that FAB-MS spectra of monensin complexes contain peaks of [MonH+Na]+ due to sodium traces from the matrix used (3-nitrobenzylalcohol), an observation that is in accordance with the high affinity of monensin to bind sodium ions and with data reported previously (Chamberlin and Agtarap 1970; Volmer and Lock 1998).

Major ions observed in the FAB-MS spectra of monensin alkaline-earth complexes 1 and 2
Cytotoxicity assay
The aerobic Gram-positive microorganisms B. subtillis, S. lutea and B. mycoides were used as test strains to evaluate the cytotoxicity of 1, 2, MgCl2.6H2O and CaCl2. The bactericidal properties of MonH were also confirmed in the present study and its determined activity is in agreement with the data previously reported (Pantcheva et al. 2008).
The alkaline-earth chlorides, similarly to MnCl2 and CoCl2 (Dorkov et al. 2008; Pantcheva et al. 2008), cannot be defined as bactericidal agents because their minimum inhibitory concentrations fall in the millimolar concentration range (Table 5). Thus the effect of metal salts is uninfluential on the visible growth of bacteria strains and can be disregarded in comparison with the corresponding metal complexes studied.
The alkaline-earth complexes of monoanionic form of monensic acid possess strongly enhanced cytotoxicity against selected strains in comparison to the free ligand (Table 5) and furthermore, the antimicrobial activity of 1 and 2 cannot be assigned to the simple additive effect of partners in the complexes. Instead, it may probably be attributed to increased penetration of the new compounds through the bacteria’s cell membrane. Previously we suggested that the increased activity of the Co2+ complex of Mon (Pantcheva et al. 2008) could be explained in the terms of an unusual coordination mode of the monovalent polyether ionophorous antibiotic. The current results manifest that the role of divalent metal ion in mononuclear complexes of deprotonated monensic acid should be reconsidered, since the coordination mode of the antibiotic remains unchanged both in transition and in alkaline-earth metal complexes, respectively, but a difference (2–7 multiple) is observed in their cytotoxic properties.
It is still early to predict the intimate antibacterial mechanism of monensin divalent metal complexes, although it seems to be closely linked to the ability of the ligand to bind metal(II) ions and to transfer them across the cell membrane as neutral complexes, followed by further dissociation of the compounds and by subsequent disturbance of homeostatic processes thus causing inhibition of bacteria growth.
Conclusion
Alkaline-earth complexes of monoanionic form of monensic acid with Mg2+ and Ca2+ were isolated for the first time in solid state. The complexes are of general composition [M(Mon)2(H2O)2] (M = Mg, Ca) and consist of an alkaline-earth metal ion placed in distorted octahedral environment. Monensin ligands act in a bidentate coordination mode via their monodentately bound carboxylate- and methoxy functions positioned at the both opposite sites of monensic anions. Additionally, two water molecules form two dative bonds with the alkaline-earth metal ion completing the sixfold geometry around the metal center and stabilizing the “head-to-tail” cyclization of the ligand molecule. The cytotoxicity assay against aerobic Gram-positive bacteria shows strongly enhanced activity of alkaline-earth monensin complexes in comparison to monensic acid that is most likely due to the unusual coordination mode of the ligand although the influence of the divalent metal ion should be also taken into account.
Supplementary data
CCDC 734913 (1) and 734912 (2) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via http://www.ccdc.cam.ac.uk (or an application from Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44 01223 336033; or e-mail: deposit@ccdc.cam.ac.uk.
References
Agtarap A, Chamberlin JW, Pinkerton M, Steinrauf LK (1967) The structure of monensic acid, a new biologically active compound. J Am Chem Soc 89:5737–5739
Ajaz AA, Robinson JA, Turner DL (1987) Biosynthesis of the polyether ionophore antibiotic monensin A: assignment of the carbon-13 and proton n. m. r. spectra of monensin A by two-dimensional spectroscopy. Incorporation of oxygen-18 labeled molecular oxygen. J Chem Soc Perkin Trans 1:27–36
Andrews JM (2001) Determination of minimum inhibitory concentrations. J Antimicrob Chemother 48:5–16
Augustine PC, Smith CK, Danforth HD, Ruff MD (1987) Effect of ionophorous anticoccidials on invasion and development of Eimeria: comparison of sensitive and resistant isolates and correlation with drug uptake. Poultry Sci 66:960–965
Augustine PC, Watkins KL, Danforth HD (1992) Effect of monensin on ultrastructure and cellular invasion by the turkey coccidia Eimeria adenoeides and Eimeria meleagrimitis. Poultry Sci 71:970–978
Berg HH, Hamill RL (1978) The isolation and characterization of narazin, a new polyether antibiotic. J Antibiot 31:1–6
Briggs RW, Hinton JF (1978) Thallium-205 and proton nuclear magnetic resonance investigation of the complexation of thallium by the ionophores monensin and nigericin. Biochemistry 17:5576–5578
Chamberlin JW, Agtarap A (1970) Observations on the mass spectrometry of monensin and related compounds. Org Mass Spectrom 3:271–285
Chappe LR (1979) The site of action of the anticoccidial salinomycin (Coxistac). J Parasitol 65:137–143
Cox BG, Vantruong N, Rzeszotarska J, Schneider H (1984) Rates and equilibria of alkali metal and silver ion complex formation with monensin in ethanol. J Am Chem Soc 106:5965–5969
Dassie SA, Baruzzi AM (2002) Alkaline-earth cation transfer assisted by monensin across the water vertical bar 1,2-dichloroethane interface in the presence of alkali cations. J Electroanal Chem 522:158–166
Dorkov P, Pantcheva IN, Sheldrick WS, Mayer-Figge H, Petrova R, Mitewa M (2008) Synthesis, structure and antimicrobial activity of manganese(II) and cobalt(II) complexes of the polyether ionophore antibiotic sodium monensin A. J Inorg Biochem 102:26–32
Duax WL, Smith GD, Strong PD (1980) Complexation of metal ions by monensin—crystal and molecular structure of hydrated and anhydrous crystal forms of sodium monensin. J Am Chem Soc 102:6725–6729
Folz SD, Lee BL, Nowakowski LH, Conder GA (1988) Anticoccidial evaluation of halofuginone, lasalocid, maduramicin, monensin and salinomycin. Vet Parasitol 28:1–9
Garcla-Rosas J, Schneider H, Cox BG (1983) Silver complexation by the ionophorous antibiotic monensin in nonaqueous solvents. J Phys Chem 87:5467–5472
Gertenbach PG, Popov AI (1975) Solution chemistry of monensin and its alkali-metal ion complexes—potentiometric and spectroscopic studies. J Am Chem Soc 97:4738–4744
Hamidinia SA, Shimelis OI, Tan B, Erdahl WL, Chapman CJ, Renkes GD, Taylor RW, Pfeiffer DR (2002) Monensin mediates a rapid and selective transport of Pb2+—possible application of monensin for the treatment of Pb2+ intoxication. J Biol Chem 277:38111–38120
Hamidinia SA, Tan B, Erdahl WL, Chapman CJ, Taylor RW, Pfeiffer DR (2004) The ionophore nigericin transports Pb2+ with high activity and selectivity: a comparison to monensin and ionomycin. Biochemistry 43:15956–15965
Hebrant M, Mimouni M, Tissier M, Pointud Y, Juillard J, Dauphin G (1992) Complexing ability of ionophore monensin for divalent cations. New J Chem 16:999–1008
Huczynski A, Michalak D, Przybylski P, Brzezinski B, Bartl F (2006a) Monensin A benzyl ester and its complexes with monovalent metal cations studied by spectroscopic, mass spectrometry and semiempirical methods. J Mol Struct 797:99–110
Huczynski A, Przybylski P, Brzezinski B (2006b) Complexes of monensin A methyl ester with Mg2+, Ca2+, Sr2+, Ba2+ cations studied by electrospray ionization mass spectrometry and PM5 semiempirical method. J Mol Struct 788:176–183
Huczynski A, Przybylski P, Brzezinski B, Bartl F (2006c) Monensin A methyl ester complexes with Li+, Na+ and K+ cations studied by ESI–MS, 1H- and 13C-NMR, FTIR, as well as PM5 semiempirical method. Biopolymers 81:282–294
Huczynski A, Przybylski P, Brzezinski B, Bartl F (2006d) Spectroscopic, mass spectrometry, and semiempirical investigation of a new ester of monensin A with ethylene glycol and its complexes with monovalent metal cations. Biopolymers 82:491–503
Huczynski A, Przybylski P, Brzezinski B (2007a) NMR, FTIR, ESI–MS and semiempirical study of a new 2-(2-hydroxyethoxy)ethyl ester of monensin A and its complexes with alkali metal cations. Tetrahedron 63:8831–8839
Huczynski A, Przybylski P, Schroeder G, Brzezinski B (2007b) Investigation of complex structures of a new 2-hydroxyethyl ester of monensin A with Mg2+, Ca2+, Sr2+, Ba2+ cations using electrospray ionization mass spectrometry and semiempirical PM5 methods. J Mol Struct 829:111–119
Huczynski A, Ratajczak-Sitarz M, Katrusiak A, Brzezinski B (2007c) Molecular structure of the 1:1 inclusion complex of monensin A lithium salt with acetonitrile. J Mol Struct 871:92–97
Huczynski A, Brzezinski B, Bartl F (2008a) Structures of complexes of benzyl and allyl esters of monensin A with Mg2+, Ca2+, Sr2+, Ba2+ cations studied by ESI–MS and PM5 methods. J Mol Struct 886:9–16
Huczynski A, Lowicki D, Brzezinski B, Bartl F (2008b) Spectroscopic, mass spectrometry and semiempirical investigation of a new 2-methoxyethyl ester of monensin A and its complexes with Li+, Na+ and K+ cations. J Mol Struct 874:89–100
Huczynski A, Lowicki D, Brzezinski B, Bartl F (2008c) Spectroscopic, mass spectrometry, and semiempirical investigations of a new 2-(2-methoxyethoxy)ethyl ester of monensin A and its complexes with monovalent cations. J Mol Struct 879:14–24
Huczynski A, Ratajczak-Sitarz M, Katrusiak A, Brzezinski B (2008d) Molecular structure of rubidium six-coordinated dihydrate complex with monensin A. J Mol Struct 888:224–229
Johnson SM, Herrin J, Liu SJ, Paul IC (1970a) Crystal structure of a barium complex of antibiotic X-537A, Ba(C34H53O8)2·H2O. J Chem Soc D Chem Commun 2:72–73
Johnson SM, Herrin J, Liu SJ, Paul IC (1970b) Crystal and molecular structure of the barium salt of an antibiotic containing a high proportion of oxygen. J Am Chem Soc 92:4428–4435
Kevin DA II, Meujo DAF, Hamann MT (2009) Polyether ionophores: broad-spectrum and promising biologically active molecules for the control of drug resistant bacteria and parasites. Expert Opin Drug Discov 4:109–146
Koinarski V, Sherkov SN (1987) Effect of anticoccidial preparations in the prevention of coccidiosis in turkeys caused by Eimeria adenoides. Vet Med Nauki 24:81–85
Liu WC, Slusarchyk DS, Astle G, Trejo WH, Brown NE, Meyers E (1978) Ionomycin, a new polyether antibiotic. J Antibiot 9:815–819
Long PL, Jeffers TK (1982) Studies on the stage of action of ionophorous antibiotics against Eimeria. J Parasitol 68:363–371
Lutz WK, Winkler FK, Dunitz JD (1971) Crystal structure of the antibiotic monensin: similarities and differences between free acid and metal complex. Helv Chim Acta 54:1103–1108
Martinek T, Riddell FG, Wilson C, Weller CT (2000) The conformations of monensin A metal complexes in solution determined by NMR spectroscopy. J Chem Soc Perkin Trans 2:35–41
Mimouni M, Pointud Y, Juillard J (1994) Interactions in methanol of divalent metal cations with bacterial ionophores, lasalocid and monensin—thermodynamical aspects. Bull Soc Chim Fr 131:58–65
Mimouni M, Hebrant M, Dauphin G, Juillard J (1996) Monovalent cation salts of the bacterial ionophore monensin in methanol. Structural aspects from NMR experiments. J Chem Res 6:278–279
Pangborn W, Duax WL, Langs D (1987) The hydrated potassium complex of the ionophore monensin A. J Am Chem Soc 109:2163–2165
Pantcheva IN, Mitewa M, Sheldrick WS, Oppel IM, Zhorova R, Dorkov P (2008) First divalent metal complexes of the polyether ionophore monensin A: X-ray structures of [Co(Mon)2(H2O)2] and [Mn(Mon)2(H2O)2] and their bactericidal properties. Curr Drug Discov Technol 5:154–161
Pantcheva IN, Dorkov P, Atanasov VN, Mitewa M, Shivachev BL, Nikolova RP, Mayer-Figge H, Sheldrick WS (2009) Crystal structure and properties of the copper(II) complex of sodium monensin A. J Inorg Biochem. doi: 10.1016/j.jinorgbio.2009.08.007
Paz FAA, Gates PJ, Fowler S, Gallimore A, Harvey B, Lopes NP, Stark CBW, Staunton J, Klinowski J, Spencer JB (2003) Sodium monensin dihydrate. Acta Cryst E59:m1050–m1052
Riddell FG (2002) Structure, conformation, and mechanism in the membrane transport of alkali metal ions by ionophoric antibiotics. Chirality 14:121–125
Rzeszotarska J, Wagner-Czauderna E, Kalinowski MK (1994) Complexation of Tl+ ions by monensin anion in binary mixtures of dipolar aprotic solvents. J Chem Res 10:400–401
Sheldrick G (1990) SHELXS-97. Program for crystal structure solution. Institut für Anorganische Chemie der Universität, Göttingen, Germany
Sheldrick G (1997) SHELXL-97. Program for crystal structure refinement. Institut für Anorganische Chemie der Universität, Göttingen, Germany
Stern PH (1977) Ionophores: chemistry, physiology and potential applications to bone biology. Clin Orthop Relat Res 122:273–298
Stiles MK, Craig ME, Gunnel SL, Pfeiffer DR, Taylor DR (1991) The formation constants of ionomycin with divalent cations in 80% methanol/water. J Biol Chem 266:8336–8342
Varga I, Sreter T (1996) Efficacy of a monensin-duokvin combination against Eimeria acervulina in chickens. Folia Parasitol (Praha) 43:153–155
Volmer DA, Lock CM (1998) Electrospray ionization and collision-induced dissociation of antibiotic polyether ionophores. Rapid Commun Mass Spectrom 12:157–164
Wagner-Czauderna E, Rzeszotarska J, Orlowska E, Kalinowski MK (1997) Complexing ability of monensin anion for alkali metal cations in binary mixtures of dipolar aprotic solvents. Phys Chem Chem Phys 101:1154–1157
Walba DM, Hermsmeier M, Haltiwanger RC, Noordik JH (1986) Crystal structures of monensin B lithium and silver salts. J Org Chem 51:245–247
Wang Z, Suo X, Xia X, Shen J (2006) Influence of monensin on cation influx and Na+ -K+ -ATPase activity of Eimeria tenella sporozoites in vitro. J Parasitol 92:1092–1096
Westley JW, Liu CM, Evans RHJr, Sello LH, Troupe N, Hermann T (1983) Preparation, properties and biological activity of natural and semisynthetic urethanes of monensin. J Antibiot 36:1195–1200
Yildirim SO, McKee V, Khardli FZ, Mimouni M, Hadda TB (2007) Rubidium(I) monensinate dihydrate. Acta Cryst E64:m154–m155
Acknowledgements
The present research is financially supported from the National Science Fund (NSF), DO-02-84/2009. SS is grateful to NSF (UNA-17/2005) for the purchase of Bruker Avance AVII+ 600 NMR spectrometer. The authors are thankful to Assoc. Prof. A. Nakov and MSci P. Dorkov (BIOVET Ltd.) for supplying sodium monensin.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pantcheva, I.N., Zhorova, R., Mitewa, M. et al. First solid state alkaline-earth complexes of monensic acid A (MonH): crystal structure of [M(Mon)2(H2O)2] (M = Mg, Ca), spectral properties and cytotoxicity against aerobic Gram-positive bacteria. Biometals 23, 59–70 (2010). https://doi.org/10.1007/s10534-009-9269-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10534-009-9269-5