
ABSTRACT
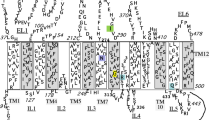
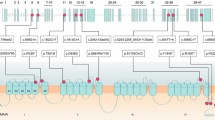
Alterations in ion channel permeability or selectivity have been shown to cause neurological defects in humans. Anion exchanger isoform 3 (AE3) is prominently expressed in the brain and performs an electroneutral exchange of chloride and bicarbonate ions. In order to study the potential role of AE3 in human neurological disease, we characterized AE3 genomic structure and performed mutational analysis on patients with an episodic movement disorder that maps to the same genetic locus. AE3 genomic organization, including the nucleotide sequence of the 5′-untranslated region and intron/exon boundaries, is highly conserved between humans and homologs from mouse and rat. Mutational analysis revealed no disease-causing defect in patients with familial paroxysmal dyskinesia, although several benign polymorphisms were identified. AE3 variation may prove useful for further genetic studies, such as finer resolution mapping. Characterization of genomic structure will facilitate mutational analysis of AE3 in studies of neurological diseases mapped to the same locus.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Accepted: May 12, 1998
Rights and permissions
About this article
Cite this article
Einum, D., Zhang, J., Arneson, P. et al. Genomic structure of human anion exchanger 3 and its potential role in hereditary neurological disease. Neurogenetics 1, 289–292 (1998). https://doi.org/10.1007/s100480050043
Issue Date:
DOI: https://doi.org/10.1007/s100480050043