
Abstract
The preliminary cytotoxic effect of 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride (1)—a potent topoisomerase II inhibitor—was measured using a MTT assay. It was found that the compound decreased the number of viable cells in both estrogen receptor-positive MCF-7 and estrogen receptor-negative MDA-MB-231breast cancer cells, with IC50 values of 146 ± 2 and 132 ± 2 μM, respectively. To clarify the molecular basis of the inhibitory action of 1, molecular docking studies were carried out. The results suggest that 1 targets the ATP binding pocket.

4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride
Similar content being viewed by others
Introduction
An important chemotherapeutic target in the treatment of cancer is the ATP-dependent enzyme topoisomerase II (Topo II), which regulates the conformational changes in DNA topology necessary for transcription, replication, and chromosome condensation and segregation [1, 2]. Currently, six Topo II inhibitors (etoposide, teniposide, doxorubicin, daunorubicin, idarubicin, and mitoxantrone) are prescribed as highly anti-neoplastic drugs in clinical use. However, emerging tumor resistance and several side effects, such as hematological toxicity, nausea and vomiting, and hair loss, are associated with them [3, 4]. Therefore, efforts have been made by many research groups to find new chemicals with improved bioactivity. Although many compounds were found as cytostatic agents and Topo II and(or) I inhibitors, most showed significant toxicity for normal dividing cells, which precludes their use as drugs or lead molecule candidates [5]. The discovery of new types of Topo II inhibitors that can be synthesized easily, show increased sensitivity in drug resistant tumors and decreased dose-limiting toxicities would be a significant addition to the choices available in the treatment of cancer.
Recently, as part of a search for the molecular basis of antibacterial activity of thiosemicarbazide-based compounds, we documented for the first time that one of the mechanisms of the bioactivity of these molecules is connected with inhibition of bacterial Topo IV [6–8]. Since both bacterial and human topoisomerases share a unique structural ATP-binding motif, called the Bergerat fold [9], we decided to conduct enzymatic studies to determine the effect of thiosemicarbazide derivatives on human topoisomerases. We found that the title compound, 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride (1) selectively inhibited Topo II activity almost completely at 15 μM concentration, proving more potent than etoposide under the same experimental conditions [10]. To expand our initial findings with further details on the biological action pathways of 1, the compound was further assessed by preliminary cytotoxicity measurements. Herein we present the results of these investigations, as well as those of subsequent docking studies and DFT calculations, which allowed us to suggest that compound 1 targets the ATP binding pocket.
Materials and methods
Etoposide and camptothecin were purchased from TopoGEN (http://www.topogen.com/). Stock cultures of breast cancer MCF-7 and MDA-MB-231 were purchased from the American Type Culture Collection (Rockville, MD). Dulbecco’s minimal essential medium (DMEM) and foetal bovine serum (FBS) used in cell culture were products of Gibco (http://www.invitrogen.com/site/us/en/home/brands/Gibco.html). Glutamine, penicillin and streptomycin were obtained from Quality Biologicals (Gaithersburg, MD). [3H]-Thymidine (6.7 Ci/mmol) was the product of NEN (Boston, MA).
Cell culture
Human breast cancer MDA-MB-231 and MCF-7 cells maintained in DMEM supplemented with 10 % fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin at 37 °C. Cells were cultured in Costar flasks and subconfluent cells were detached with 0.05 % trypsin and 0.02 % EDTA in calcium-free phosphate buffered saline, counted in hemocytometers and plated at 5 × 105 cells per well in six-well plates (Nunc, http://www.nuncbrand.com) in 2 ml of growth medium (DMEM without phenol red with 10 % CPSR1). Cells reached about 80 % of confluency at day 3 and in most cases such cells were used for the assays.
Cytotoxic assay
To examine the effect of the studied drugs on MCF-7 and MDA-MB-231cell proliferation, the cells were seeded in 24-well tissue culture dishes at 1 × 105 cells/well with 1 ml growth medium. After 48 h (1.8 ± 0.1 × 105 cells/well) plates were incubated with varying concentrations of compound 1, chlorambucil and 0.5 μCi [3H]-thymidine for 24 h at 37 °C. Cells were rinsed three times with PBS, solubilised with 1 ml 0.1 M sodium hydroxide containing 1 % SDS, scintillation fluid (9 ml) was added and radioactivity incorporation into DNA was measured in a scintillation counter.
Cell viability assay
The assay was performed according to the method of Carmichael [11] using 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma, St. Louis, MO). Confluent cells, cultured for 24 h with various concentrations of the studied compounds in six-well plates were washed three times with PBS and then incubated for 4 h in 1 ml MTT solution (5 mg/ml PBS) at 37 °C in 5 % CO2 in an incubator. The medium was removed and 1 ml 0.1 M HCl in absolute isopropanol was added to attached cells. Absorbance of converted dye in living cells was measured at a wavelength of 570 nm. Cell viability of breast cancer cells cultured in the presence of ligands was calculated as a per cent of control cells.
Automated docking setup
Docking was performed by means of the FlexX program [12] as implemented in LeadIT software package [13] using models of Topo II binding site complexed with AMP-PNP (PDB id 1ZXN [14]) and etoposide (PDB id 3QX3 [15]) as templates. The ligand within the active site and all water molecules were removed while magnesium ion was allowed to remain with the charge of +2. The active site was defined to include all atoms within 6.5 Å radius of the native ligand. The first 100 top ranked docking poses were saved for each docking run. Subsequently, compounds 1–3 were docked using same docking parameters. Both rigid and flexible docking was performed (the results for the latter are presented). For compounds 1 and 3, both piperidine protonated and deprotonated forms were considered.
DFT calculations
Following the methodology used previously [16] energies of best poses of compounds 1–4 obtained from FlexX docking were calculated using the B3LYP functional [17, 18] expressed in the basis set of 6–31 G(d) [19–21] as implemented in Gaussian 09 [22] to describe their molecular properties. Natural population analysis (NPA) phase of NBO was used [23]. The highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals were illustrated using the GaussView 5.0 program [24].
Statistical analysis
In all experiments, the mean values for six independent experiments ± standard deviations (SD) were calculated, unless otherwise indicated. The results were submitted to statistical analysis using Students t-test, accepting P < 0.05 as significant.
Results and discussion
The titled 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride, 1, was synthesized through the route outlined in Fig. 1; details of the synthetic procedure, structural characterization of 1 and its effect on human topoisomerase II activity are described in our patent application [10].

Synthetic route for 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride (1)
Cytotoxic effect of 1
Many reports in recent years have indicated that inhibitory action towards Topo II coincides with cytotoxic properties for several naturally occurring and synthetic compounds [25–30]. We therefore subjected 1 to a preliminary MTT assay in estrogen receptor-positive (MCF-7) and estrogen receptor-negative (MDA-MB-231) breast cancer cells to identify any possible correlation. Compound 1 decreased the number of viable cells in both MCF-7 and MDA-MB-231 breast cancer cells; however, at somewhat higher concentrations than the positive control chlorambucil. The inhibitory activities (IC50) of compound 1 on MDA-MB-231 and MCF-7 were 146 ± 2 and 132 ± 2 μM, respectively, whereas that of chlorambucil were 92 ± 2 and 97 ± 2 μM, respectively, as shown in Fig. 2. Although the cytotoxicity of 1 was concentration-dependent in both cell lines, it was more pronounced at shorter times in MDA-MB-231 than in MCF-7.

Viability of MCF-7 (a) and MDA-MB-231 (b) cells treated for 24 h with different concentrations of compound 1 and chlorambucil. Mean values ± SD from three independent experiments performed in duplicate are presented
The above data was corroborated by a cell proliferation assay. The profiles of DNA synthesis were found to be similar in MCF-7 and MDA-MB-231 (Fig. 3). The concentration of 1 required to inhibit [3H]-thymidine incorporation into DNA by 50 % (IC50) in MDA-MB-231 was found to be 123 ± 2 μM, suggesting a lower cytotoxic potency compared to chlorambucil (IC50 = 56 ± 2 μM). The concentrations of 1 and chlorambucil required for 50 % inhibition of [3H]-thymidine incorporation into DNA in breast cancer MCF-7 cells (IC50) were 124 ± 2 μM and 65 ± 2 μM, respectively.

Cytotoxic effects of compound 1 and chlorambucil on the cultured breast cancer cell lines MDA-MB-231 (a) and MCF-7 (b) as measured by inhibition of [3H]-thymidine incorporation into DNA. Mean values ± SD of three independent experiments performed in duplicate are presented
Computational studies
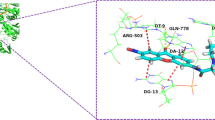
To examine the mode of inhibitory action of 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride, 1, docking simulations were performed with ATP-binding domain of hTopoIIα (PDB: 1ZXM) and DNA binding site of hTopoIIβ (PDB id: 3QX3) using the FlexX program, which has been shown to be reliable enough to carry out binding mode analysis of ligands in ATP binding pocket of hTopoIIα [31]. To validate the molecular docking protocol, AMPPNP and etoposide (bound ligands) were initially docked into the crystal structure of the enzyme. The docked ligands were found to have similar binding poses to the co-crystallized ligand, thus validating the adopted docking methodology. We then docked 1 into the DNA and ATP binding sites, respectively, and its interactions with the target enzyme were analyzed. In both cases, crystallographic water molecules were treated rotatable and displaceable. Their contribution to binding was found negligible in all cases. Free energies of binding of 1, obtained from the Hyde scoring function [32, 33], are 9 kJ/mol in the DNA active site while it is −20 kJ/mol in the ATP binding site (vs −6 kJ/mol for AMPPNP, 2, an analog of ATP crystallized with hTopoIIα). From this data we conclude that 1 has greater affinity toward the ATP binding site than to the DNA binding pocket of hTopoIIα and is an even more effective inhibitor than AMPPNP. Figures 4 and 5 show interactions of 1 in DNA and ATP active sites. As can be seen, compound 1 fits well into the ATP pocket, forming 11 hydrogen bonding interactions with water molecules, Gly164, Asn163, Arg162, Glu87, Gly166, Tyr165 and hydrophobic interactions with Gly160, Gly161, Asn91.

Interactions between 1 and the residues of the DNA-(left) and ATP-(right) binding sites

Docking results with ATP-binding domain of hTopoIIα (PDB: 1zxm). Left Superposition of the native ligand AMPPNP (rendered as tubes), and the best conformation of 1. Right Binding mode of 1
To test the docking predictions, we repeated the docking simulations with the closely related structural analog of 1, 4-(4-methoxyphenyl)-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride 3 (Fig. 6), which was found to be inactive in our enzyme bioassays. Consistent with these results, the docking calculations correctly predict lack of stability of the active site—compound 3 complex (27 kJ/mol), confirming the reliability of the employed docking methodology.

The chemical structure of 4-(4-methoxyphenyl)-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide hydrochloride (3), which was found to be inactive as hTopo II inhibitor
Most recently, some meaningful results concerning the ligand recognition process in the ATP binging site were described by Ma and co-workers [16]. In this latter communication, the authors applied a combination of molecular docking approaches, DFT calculations, and CoMFA/CoMSIA methods to explore the possible binding modes of hTopo II inhibitors with the naphthoquinone core structure. Based on the best-scored conformations obtained from the FlexX docking simulation they generated the distribution of frontier molecular orbitals and found significant differences between potent and weak inhibitors in their HOMO distribution. The HOMO of the potent inhibitors covered almost the whole molecule while weak inhibitors had their HOMO outside the phenyl ring. It was thus proposed that distribution of HOMO significantly affects the binding of ligands to the ATP pocket. Following these findings, and using the same level of theory, we generated the HOMO orbitals for 1 and 3 and compared them with HOMO of the potent hTopoIIα catalytic inhibitor (which we refer to as 4, Fig. 7) from the closely related class reported earlier in the literature [34]. Comparison of the orbitals leads to the conclusion that there is no similarity between HOMO distributions of the studied compounds and AMPPNP. A similar conclusion can be drawn from analysis of the LUMO distributions (MOs are given in Fig. S1 in the Supplementary Material). Thus it seems that the electronic distribution in the frontier orbitals is not related to inhibitory activity of studied compounds against Topo II—a conclusion in line with the lack of covalent bonding between inhibitor and active site residues.

Chemical structure of thiosemicarbazone derivative, 4—a recently identified hTopoIIα catalytic inhibitor [31]
Conclusions
In this contribution we show compound 1 to be highly selective and potent inhibitor of human Topo II. From docking simulations, we conclude that its inhibitory action is connected with the ATP binding pocket although competitive inhibition assays are needed to confirm this hypothesis. These results will aid the rational design of novel thiosemicarbazide-based compounds that target the inhibition of Topo II and will provide insights into the discovery of novel anticancer agents.
References
Kellner U, Sehested M, Jensen PB, Gieseler F, Rudolph P (2002) Culprit and victim-DNA topoisomerase II. Lancet Oncol 3:235–243
Kaufmann SH, Gore SD, Miller CB, Jones RJ, Zwelling LA, Schneider E et al (1998) Topoisomerase II and the response to antileukemic therapy. Leuk Lymphoma 29:217–237
Karki R, Thapa P, Young Yoo H, Man Kadayat T, Park P-H, Na Y, Lee E, Jeon KH, Cho W-I, Choi H, Kwonc Y, Lee ES (2012) Dihydroxylated 2,4,6-triphenyl pyridines: synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure-activity relationship study. Eur J Med Chem 49:219–228
Trojan L, Kiknavelidze K, Knoll T, Alken P, Michel MS (2005) Prostate cancer therapy: standard management, new options and experimental approaches. Anticancer Res 25:551–561
Wu W-B, Ou J-B, Huang Z-H, Chen S-B, Ou T-M, Tan J-H, Li D, Shen L-L, Huang S-L, Gu L-Q, Huang Z-S (2011) Synthesis and evaluation of mansonone F derivatives as topoisomerase inhibitors. Eur J Med Chem 46:3339–3347
Siwek A, Stączek P, Stefańska J (2011) Synthesis and structure-activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with Gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur J Med Chem 46:5717–5726
Siwek A, Stączek P, Wujec M, Stefańska J, Kosikowska U, Malm A, Jankowski S, Paneth P (2011) Biological and docking studies of topoisomerase IV inhibition by thiosemicarbazides. J Mol Model 17:2297–2303
Siwek A, Stączek P, Kosikowska U, Malm A, Paneth P, Jankowski S (2012) Does dehydrocyclization of 4-benzoylthiosemicarbazides in acetic acid lead to s-triazoles or thiadiazoles? Struct Chem 23:1441–1448. doi:10.1007/s11224-012-0065-4
Gadelle D, Graille M, Forterre P (2006) The HSP90 and DNA topoisomerase VI inhibitor radicicol also inhibits human type II DNA topoisomerase. Biochem Pharmacol 72:1207–1216
Siwek A, Wujec M, Bielawska A, Bielawski K. Polish patent application P397526
Carmichael J, DeGraff W, Gazdar A, Minna J, Mitchell J (1987) Evaluation of a tetrazolium-based semiautomated colorimetric assay: Assessment of chemosensitivity testing. Cancer Res 47:936–942
Kramer B, Rarey M, Lengauer T (1999) Evaluation of the FlexX incremental construction algorithm for protein-ligand docking. Proteins 37:228–241
LeadIT 2.1.0, BioSolveIT GmbH, St. Augustin, Germany, 2012
Wei H, Ruthenburg AJ, Bechis SK, Verdine GL (2005) Nucleotide-dependent domain movement in the ATPase domain of a human type IIA DNA topoisomerase. J Biol Chem 280:37041–37047
Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW, Chan NL (2011) Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333:459–462
Ma Y, Wang J-G, Wang B, Li Z-M (2011) Integrating molecular docking, DFT and CoMFA/CoMSIA approaches for a series of naphthoquinone fused cyclic α-aminophosphonates that act as novel topoisomerase II inhibitors. J Mol Model 17:1899–1909
Becke AD (1993) Density–functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993(98):5648–5652
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti conelation energy formula into a functional of the electron density. Phys Rev B 37:785–789
Ditchfield R, Hehre WJ, Pople JA (1971) Self–consistent molecular–orbital methods. IX. An extended Gaussian–type basis for molecular–orbital studies of organic molecules. J Chem Phys 54:724–728
Francl M, Pietro WJ, Hehre WJ, Binkley JS, Gordon MS, DeFrees DJ, Pople JA (1982) Self–consistent molecular orbital methods. XXIII. A polarization–type basis set for second–row elements. J Chem Phys 77:3654–3665
Frisch MJ, Pople JA, Binkley JS (1984) Self–consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J Chem Phys 80:3265–3269
Frisch MJ et al (2009) Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT
Foster JP, Weinhold F (1980) Natural hybrid orbitals. J Am Chem Soc 102:7211–7218
Frisch Æ, Hratchian HP, Dennington RD II, Todd A, Keith TA, Millam J (2009) GaussView 5. Gaussian, Wallingford, CT
Zhao Y, Ge CW, Wu ZH, Wang CN, Fang YH, Zhu L (2011) Synthesis and evaluation of aroylthiourea derivatives of 4-β-amino-4′-O-demethyl-4-desoxypodophyllotoxin as novel topoisomerase II inhibitors. Eur J Med Chem 46:901–906
Shi DK, Zhang W, Ding N, Li M, Li Y-X (2012) Design, synthesis and biological evaluation of novel glycosylated diphyllin derivatives as topoisomerase II inhibitors. Eur J Med Chem 47:424–431
Han S, Xiao Z, Bastow KF, Lee K-H (2004) Antitumor agents. Part 230: C4′-esters of GL-331 as cytotoxic agents and DNA topoisomerase II inhibitors. Bioorg Med Chem Lett 14:2979–2982
Shinkre BA, Raisch KP, Fan L, Velu SE (2007) Analogs of the marine alkaloid makaluvamines: synthesis, topoisomerase II inhibition, and anticancer activity. Bioorg Med Chem Lett 17:2890–2893
Woo S, Jung J, Lee C, Kwon Y, Na Y (2007) Synthesis of new xanthone analogues and their biological activity test - Cytotoxicity, topoisomerase II inhibition, and DNA cross-linking study. Bioorg Med Chem Lett 17:1163–1166
Joseph B, Facompré M, Da Costa H, Routier S, Mérour JY, Colson P, Houssier C, Bailly C (2001) Synthesis, cytotoxicity, DNA interaction and topoisomerase II inhibition properties of tetrahydropyrrolo[3,4-a]carbazole-1,3-dione and tetrahydropyrido-[3,2-b]pyrrolo[3,4-g]indole-1,3-dione derivatives. Bioorg Med Chem 9:1533–1541
Baviskar AT, Madaan C, Preet R, Mohapatra P, Jain V, Agarwal A, Guchhait SK, Kundu CN, Banerjee UC, Bharatam PV (2011) N-fused imidazoles as novel anticancer agents that inhibit catalytic activity of topoisomerase IIα and induce apoptosis in G1/S phase. J Med Chem 54:5013–5030
Schneider N, Hindle S, Lange G, Klein R, Albrecht J, Briem H, Beyer K, Claußen H, Gastreich M, Lemmen Ch, Rarey M Substantial improvements in large-scale redocking and screening using the novel HYDE scoring function. J Comput Aided Mol Des 26:701–723. doi: 10.1007/s10822-011-9531-0.
Reulecke I, Lange G, Albrecht J, Klein R, Rarey M (2008) Towards an integrated description of hydrogen bonding and dehydration: Decreasing false positives in virtual screening with the HYDE scoring function. Chem Med Chem 3:885–897
Huang H, Chen Q, Ku X, Meng L, Lin L, Wang X, Zhu C, Wang Y, Chen Z, Li M, Jiang H, Chen K, Ding J, Liu H (2010) A series of alpha-heterocyclic carboxaldehyde thiosemicarbazones inhibit topoisomerase IIalpha catalytic activity. J Med Chem 53:3048–3064
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 2533 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Siwek, A., Stączek, P., Wujec, M. et al. Cytotoxic effect and molecular docking of 4-ethoxycarbonylmethyl-1-(piperidin-4-ylcarbonyl)-thiosemicarbazide—a novel topoisomerase II inhibitor. J Mol Model 19, 1319–1324 (2013). https://doi.org/10.1007/s00894-012-1679-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00894-012-1679-6