
Abstract
Tyramine β-monooxygenase (TBM) is a member of a family of copper monooxygenases containing two noncoupled copper centers, and includes peptidylglycine monooxygenase and dopamine β-monooxygenase. In its Cu(II) form, TBM is coordinated by two to three His residues and one to two non-His O/N ligands consistent with a [CuM(His)2(OH2)2–CuH(His)3(OH2)] formulation. Reduction to the Cu(I) state causes a change in the X-ray absorption spectroscopy (XAS) spectrum, consistent with a change to a [CuM(His)2S(Met)–CuH(His)3] environment. Lowering the pH to 4.0 results in a large increase in the intensity of the Cu(I)–S extended X-ray absorption fine structure (EXAFS) component, suggesting a tighter Cu–S bond or the coordination of an additional sulfur donor. The XAS spectra of three variants, where the CuM Met471 residue had been mutated to His, Cys, and Asp, were examined. Significant differences from the wild-type enzyme are evident in the spectra of the reduced mutants. Although the side chains of His, Cys, and Asp are expected to substitute for Met at the CuM site, the data showed identical spectra for all three reduced variants, with no evidence for coordination of residue 471. Rather, the K-edge data suggested a modest decrease in coordination number, whereas the EXAFS indicated an average of two His residues at each Cu(I) center. These data highlight the unique role of the Met residue at the CuM center, and pose interesting questions as to why replacement by the cuprophilic thiolate ligand leads to detectable activity whereas replacement by imidazole generates inactive TBM.
Similar content being viewed by others

Introduction
Tyramine β-monooxygenase (TBM) is a member of a small class of copper-containing monooxygenases which utilize a pair of mononuclear copper centers at their active sites [1–4] to catalyze the hydroxylation of benzylic or peptidylglycyl Cα carbon atoms. Other members of the family include dopamine β-monooxygenase (DBM; catecholamine biosynthesis), peptidylglycine monooxygenase (PHM; C-terminal peptide amidation), and monooxygenase X (unknown function) [5]. Akin to the mammalian enzymes DBM and PHM, TBM is associated with neurotransmission in invertebrates, and catalyzes the hydroxylation of tyramine to octopamine (Fig. 1b); both molecules are critical to physiological functions, such as neuromuscular transmission and behavioral development, in insects [6–8]. Whereas a significant database of spectroscopic information has been accumulated for these enzymes [9–13], crystallographic characterization has only been achieved for PHM [14, 15]. The two copper centers of the monooxygenases (denoted CuM and CuH) are bound in a solvent-filled cleft approximately 11 Å apart, and are structurally and spectroscopically distinct. CuM is coordinated by two His residues and a Met residue, whereas CuH is coordinated to three His residues. A structure of the reduced enzyme co-crystallized with a slow substrate has revealed the presence of a dioxygen molecule bound at the CuM center, where the O–O bond length is suggestive of a Cu(II)–superoxo complex [16]. This observation, together with further kinetic [17, 18], biochemical [19], and theoretical [20] evidence, has led to the proposal that the active oxygen species is best formulated as a Cu(II)–superoxo species.

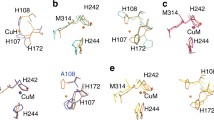
a The H and M centers of the mononuclear monooxygenases modeled on the crystal structure (Protein Data Bank file 3PHM) of the catalytic core of peptidylglycine monooxygenase. b Reaction catalyzed by tyramine β-monooxygenase. c Modeling of His, Asp, and Cys at residue 471 into the catalytic M-site showing the potential for coordination of each ligand at the CuM center
The Met residue at the CuM center appears to play a critical but as yet elusive role in catalysis. Early studies on PHM M314X substitutions reported undetectable catalytic activity in spent medium from PHM cell lines expressing the Ile, His, Asp, and Cys variants [21, 22]. More recent studies using purified variants of TBM in which the homologous Met ligand (M471) was mutated to His, Asp, or Cys have established a dramatic decrease in activity, with only the M471C variant displaying measurable activity [9]. Extended X-ray absorption fine structure (EXAFS) studies on both DBM [10, 11] and PHM [12, 23, 24] have determined that the sulfur from the Met residue is a ligand to CuM in the reduced but not the oxidized enzyme. However, a crystal structure of the M314I mutant of PHM revealed changes at both CuM and CuH suggestive of a more complex role than simple copper coordination [25]. Model studies on mononuclear Cu(II)–superoxo complexes have in general provided limited insight into the contribution of a coordinated thioether to oxygen activation [26]. The thioether moiety has little effect on the reactivity of the synthetic copper complexes. However, incorporation of thioether into a mononuclear β-diketonate Cu(II) superoxo species suggested complex chemistry involving fluxional Met coordination at low temperature [27, 28], which complemented observations from EXAFS that the PHM Cu–S(Met) bond is unusually weak, and also possibly fluxional. This led to the suggestion that the dynamics of the Cu–S(Met) interaction in PHM may play a role in providing a vibrational coordinate for hydrogen-atom tunneling during hydrogen-atom abstraction from the peptide substrate [24].
Other explanations for the important role of the M-site Met residue in catalysis have been advanced. It has been suggested that the weak donor power of the Met residue [17] prevents the copper–dioxygen complex from undergoing significant O2 reduction prior to substrate activation by hydrogen-atom abstraction. In essence, this effect would shift the equilibrium for Cu(I)–dioxygen binding to the left, by S(Met) stabilization of the Cu(I) form, thereby inhibiting uncoupling reactions induced by superoxide or peroxide “leakage” from the active site [17, 20]. A Cu(I) stabilizing role of Met has been observed experimentally in studies of the peroxide reactivity of PHM where analysis of product isotopomers clearly showed an equilibrium between Cu(II)–superoxo and Cu(I)–dioxygen species [19]. However, despite much work, the role of the thioether ligand in catalysis remains a major unanswered question.
The expression of TBM in Drosophila Schneider 2 (S2) cells [4, 29] provides a useful platform for mutagenesis studies to further probe the role of the Met residue at CuM in catalysis. Recently copper binding and enzymatic activity data were reported for a series of mutants at the M471 residue in TBM, including the Met to Cys, His, and Asp variants [9]. As stated previously, only the Cys variant retained measurable catalytic activity. The oxidized Cu(II) forms of all three variants bound an equivalent amount of copper, and showed no perturbation of their EPR spectral properties relative to the wild-type protein. In the present study, EXAFS measurements on these isolated TBM mutants have allowed us to examine, for the first time, potential structural correlations to the altered reactivities. Since the Met residue forms only a weak axial interaction in the oxidized form of PHM [13, 23] and DBM [10], major differences in copper coordination are only anticipated in the reduced forms. Here, we used X-ray absorption spectroscopy (XAS) to probe the structure of the copper centers in the wild type and each of the M471 variants, and in particular to assess whether residue 471 is able to bind to the reduced copper center. Changes in the Cu(I) coordination sphere of the mutant enzyme forms would have significant implications for the reaction of the enzyme with dioxygen, and for the subsequent formation of relevant intermediates. Although wild-type TBM appears to bind M471 in a fashion similar to PHM and DBM, we find no spectroscopic evidence for strong interactions of His, Asp, or Cys with the CuM center.
Materials and methods
Materials
Drosophila S2 cells, insect cell growth media, and Drosophila Expression System were purchased from Invitrogen. Blasticidin was purchased from Sigma. Primers were custom-ordered, high-performance-liquid-chromatography-purified, from Operon. Chromatography media was purchased from GE Healthcare. All other materials were obtained from Sigma.
Protein expression
For the purposes of the XAS studies, wild-type and mutant TBM lacking the His-tag were used. A stop codon was introduced into the pBipTBM plasmid DNA for wild-type TBM and TBM mutants [9], preceding the sequence of bases encoding for the rTEV recognition site and His-tag contained in the original construct [4]. The altered pBipTBM plasmid was transformed into Escherichia coli strain XL1 Blue (Stratagene) cells and purified using a QIAGEN HiSpeed plasmid midi kit. The composition of the purified plasmid was confirmed by DNA sequencing (University of California, Berkeley, Sequencing Facility), prior to transfection into the S2 cells. The expression of wild-type and mutant TBM in Drosophila S2 cells was performed according to previously described procedures [4].
Protein purification
All purification steps were carried out at 277 K. The recombinant enzyme was purified from the culture medium as follows. The cell culture (1.5 L) was centrifuged (3,000 rpm, 10 min) and the supernatant batch-bound to 200 mL (diethylamino)ethyl–Sepharose Fast Flow resin (10 mM potassium phosphate, pH 8), as previously described [4]. The column-bound protein was washed with 2 L of equilibration buffer, and the protein was subsequently eluted with a buffer gradient (1 L, 10 mM to 0.25 M potassium phosphate, pH 8). Protein-containing fractions (as determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis) were combined, dialyzed overnight (10 kDa cutoff membrane) against 4 L of tris(hydroxymethyl)aminomethane (Tris) buffer (20 mM Tris, 50 mM NaCl, pH 7.5), concentrated to approximately 20 mL (Millipore Ultrafree centrifugal concentrators, 50 kDa cutoff membrane), and loaded onto a Q-Sepharose column (15 mL) equilibrated with the same low-salt buffer. The column was washed with 100 mL of the equilibration buffer, and the protein was eluted using a NaCl gradient (300 mL, 20 mM Tris, 0.05 M to 0.2 M NaCl; followed by 100 mL, 0.2 M to 0.5 M NaCl). The protein content of the fractions was again determined by sodium dodecyl sulfate polyacrylamide gel electrophoresis. The purest fractions were combined, concentrated to less than 1 mL, and purified using size-exclusion chromatography, as described previously [4]. Single-banded fractions were combined and used for the preparation of XAS samples.
Preparation of samples for XAS
The purified wild-type and mutant TBM samples (3–6 mg, 50 mM Tris, 0.1 M NaCl, pH 7.5) were copper-reconstituted by dialysis against a tenfold volume of the same Tris buffer containing 40 μM CuSO4, for 6 h (10 kDa cutoff membrane). The samples were subsequently concentrated to 1 mL (10 kDa cutoff membrane), diluted to 5 mL with low-copper-containing buffer (50 mM Tris, 0.1 M NaCl, 5 μM CuSO4, 24% ethylene glycol, pH 7.5), and finally reconcentrated to a volume of less than 1 mL.
The low-pH wild-type TBM sample was prepared by 12-fold dilution of a concentrated sample at pH 7.5 with low-pH Tris buffer (50 mM Tris, 0.1 M NaCl, 21% ethylene glycol, pH 4) followed by subsequent reconcentration of the sample to a final volume of less than 1 mL.
Reduced protein samples were prepared in an inert atmosphere glove box. An excess of ascorbate ( more than 4.5 equiv; the ascorbate solution was 1.5 mM in deoxygenated water) was added to a concentrated sample of wild-type or mutant TBM, prepared as described above, that had been deoxygenated by sparging it with water-saturated argon. Samples were spun down (8,000 rpm, 1 min) to remove any precipitate.
All enzyme samples for XAS contained approximately 20% ethylene glycol. Eighty microliters of each prepared solution was added to an XAS sample holder via a syringe, and frozen in liquid nitrogen. The final TBM concentration of all XAS samples ranged from 150 to 450 μM. Protein concentrations were determined both by Bradford assays and by UV–vis (A 280) spectroscopy. Molecular weights and extinctions coefficients (A 280) for wild-type and mutant TBM lacking the His-tag were determined using ExPASy, on the assumption that all Cys are half-Cys and neglecting any posttranslational glycosylation (http://www.expasy.org). The concentrations determined by Bradford assays were within 5% of the values derived from the absorbance at 280 nm.
Collection and analysis of XAS data
Copper K-edge (8.9 keV) EXAFS and X-ray absorption near edge structure (XANES) data were collected at the Stanford Synchrotron Radiation Lightsource operating at 3 GeV with currents between 100 and 80 mA. Cu(I)-containing samples were measured on beam line 9-3 using a Si[220] monochromator and a rhodium-coated mirror upstream of the monochromator with a 13 keV energy cutoff to reject harmonics. A second rhodium mirror downstream of the monochromator was used to focus the beam. Data were collected in fluorescence mode using a high-count-rate Canberra 30-element germanium array detector with maximum count rates below 120 kHz. A Z-1 nickel oxide filter and a Soller slit assembly were placed in front of the detector to reduce the elastic scatter peak. Cu(II) samples were measured on beam line 7-3 using lower fluxes and unfocused optics, but with a similar beam line configuration of monochromator and harmonic-rejection mirror. Energy calibration was achieved by reference to the first inflection point of a copper foil (8,980.3 eV) placed between the second and third ionization chambers.
The samples (80 μL) were measured as aqueous glasses (more than 20% ethylene glycol) at 10–15 K. Six scans of a sample containing only sample buffer were collected, averaged, and subtracted from the averaged data for the protein samples to remove Z-1 Kβ fluorescence and produce a flat pre-edge baseline. Data reduction and background subtraction were performed using the program modules of EXAFSPAK [30]. Data from each detector channel were inspected for glitches, dropouts, or other nonlinear behavior before inclusion in the final average.
Spectral simulation was carried out using the program EXCURVE 9.2 [31–34] as previously described [23]. EXAFS data were simulated using a mixed-shell model consisting of imidazole and S(Met) coordination. First-shell distances (R) and Debye–Waller factors (2σ 2) for the Cu–N(imidazole) and the Cu–S(Met) shell, and the threshold energy E 0 were initially refined. In these preliminary refinements, the imidazole ring outer-shell carbon and nitrogen atoms were constrained to move relative to the first-shell Cu–N distance so as to maintain the idealized ring geometry, and all single- and multiple-scattering pathways were included in the calculations as previously described [23]. Later in the refinement, this constraint was lifted, and the outer shells of the imidazole rings were allowed to float within 10% of their original idealized positions. In practice, the final outer-shell coordinates for acceptable fits deviated by less than the permitted amount from the idealized position. The parameters refined in the fit included shell occupancy N, copper-scatterer distance R, and Debye–Waller factor (2σ 2) for each shell, and the threshold energy (E 0) for photoelectron ionization, which was constrained to be the same for all shells of scatterers.
Results
XAS of the wild-type enzyme
Previous studies of copper loading to TBM showed that the enzyme bound 1.9 coppers per protein [9] and sequence homology between TBM and PHM with respect to copper-binding residues suggests these two copper centers are chemically distinct. Since X-ray absorption detects only the average ligand environment, chemical modeling is required to gain information specific to each site. In a previous EXAFS study of PHM, we used the information from the crystal structure to generate a model which refined the two copper centers independently [23], the accuracy of which was later confirmed by Chen et al. [13] using geometry optimization of the crystal coordinates by density functional theory calculations. In the present case, we applied similar methods. The data were fit initially by an average coordination over both copper centers, and were subsequently tested for improvements in goodness of fit (F) when the copper centers were modeled separately. Figure 2 compares the Fourier transforms (FT) and EXAFS spectra for the oxidized and reduced forms of TBM. The oxidized form shows a spectrum typical of Cu(II)–imidazole coordination as expected from His ligation. The data fit well to 2.5 His residues and 1.5 O/N ligands, which represent the average coordination at each copper. No improvement in the value of F was obtained by modeling the coppers individually. The EXAFS spectrum of reduced TBM differs significantly from that of the oxidized protein, showing a large decrease in amplitude of the first shell in the FT. A single shell of imidazole groups was generally sufficient to account for the EXAFS and transform features. Figure 2 also shows the fit of reduced TBM at pH 7.0, corresponding to two His residues with Cu–N distances of 1.94 Å and 0.5 sulfur at 2.24 Å (Table 1, reduced protein fit A). The Debye–Waller term for the Cu–N(His) shell is unusually large and suggests a large spread in the individual Cu–N(His) distances. This suggested the presence of two coordinatively distinct copper centers, and in this case warranted further exploration of chemical inequivalence between the copper centers. This was achieved by assuming inequivalence of the His coordination at each copper, and simulating the data with two independent shells of imidazole ligands. Splitting the imidazole scattering into two separate contributions produced no improvement in the value of the goodness-of-fit parameter F, but resulted in more reasonable values for the Debye–Waller factors of each shell (Table 1, reduced protein fit B, Fig. S1). Notably, one shell of His residues appears to be at a short distance of 1.88 Å, a distance typical of two-coordinate Cu(I)–imidazole complexes [35–37], whereas the other shell refines to R = 1.98 Å, a distance more characteristic of three-coordinate Cu(I). We assign the two- and three-coordinate sites to the H-site and M-site, respectively.

Phase-corrected Fourier transforms and extended X-ray absorption fine structure (EXAFS) (upper insets) for oxidized (top) and ascorbate-reduced (bottom) copper centers in tyramine β-monooxygenase. Solid black lines are experimental data and dashed red lines are simulations using parameters listed in Table 1. The lower insets show the X-ray absorption near edge structure (XANES) region of the spectrum
We also explored fits involving higher imidazole coordination numbers, since homology to PHM predicts that all five His residues could remain coordinated to Cu(I) in the reduced form, leading to the predicted average imidazole per copper of 2.5 [14, 15, 23]. Fits using a single shell of 2.5 His per copper gave acceptable F values but with even larger Cu–N Debye–Waller factors (2σ 2 = 0.022 Å2, Table 1, reduced protein fit C). Using the above reasoning, this implies more heterogeneity between the two Cu(I) centers, but unexpectedly, two-shell fits with increased coordination number resulted in extremely poor fits to the data, with an unacceptably larger Debye–Waller factor at one copper center. The conclusion that we draw from this analysis is that the Cu(I) centers are most likely each coordinated by only two EXAFS-detectable His ligands. However, given the difficulty in determining EXAFS-derived coordination numbers to high precision, the presence of a third His residue at the CuH center cannot be discounted.
The identity of the remaining ligands at the M-site is less clear from the EXAFS data. Inclusion of a weak-interaction 0.5 sulfur donor at 2.24 Å improves the fit by broadening the first shell in the FT sufficiently to fit the experimental data. However, the Debye–Waller factor is unusually high for an isolated absorber–scatterer interaction, and suggests there may be two or more conformations for the Met ligand. Metal-S(Met) coordination often exists as multiple conformers with the terminal methyl group of one rotated relative to the other [38]. The best fit to the data is listed in Table 1, fit A. Figure S1 compares fits using one versus two groups of imidazoles, while Figure S2 shows the effect of ommitting the sulfur ligation.
pH dependence of the reduced wild-type protein
PHM shows a pH-dependent structural transition in which the intensity of the Cu–S interaction increases with decreasing pH with a pK of approximately 4.6 in 2-morpholinoethanesulfonic acid buffer [24]. Given the homology between PHM and TBM, it was of interest to determine whether a similar structural transition was present in TBM. Figure 3 compares the FTs of reduced TBM at pH 4 and pH 7, whence it can be seen that the intensity of the Cu–S peak at R ~ 2.3 increases dramatically at pH 4. Figure 3 also shows a simulation of the low-pH data with two Cu–N(His) interactions at 1.94 Å and one Cu–S/Cl interaction at 2.28 Å. The increase in shell occupancy from 0.5 to one Cu–S/Cl suggests that an additional Cu–S/Cl interaction is observed at low pH but the correlation between shell occupancy and the Debye–Waller factor implies that the alternative of a more rigid (lower Debye–Waller factor) Cu–S interaction should also be considered. In the PHM system the increase in Cu–S intensity at low pH could be simulated either by an additional Cu–S interaction or by allowing the Debye–Waller factor to decrease at low pH, and it was argued that the latter would imply a transition from a flexible to a rigid conformation [24]. The pK for the transition correlated with the decrease in activity at low pH, and it was suggested that the active form of the enzyme therefore required a flexible conformation with a weak or fluxional Cu–S(Met) interaction at CuM. In contrast, the TBM low-pH data cannot be adequately simulated with 0.5 Cu–S and a low Debye–Waller factor. Furthermore, the bond length for the Cu–S/Cl interaction increases from 2.24 Å at pH 7 to 2.28 Å at pH 4. This increase in bond length is inconsistent with a strengthening of the Cu–S bond which is implied by a lower Debye–Waller factor, and suggests that the increased intensity more likely arises from an additional Cu–S/Cl ligation at one of the copper centers at low pH.

pH dependence of reduced wild-type (WT) tyramine β-monooxygenase. Top: Comparison of the phase-corrected Fourier transforms at pH 7 (black) and pH 4 (red). Bottom: Phase-corrected Fourier transforms and EXAFS (upper inset) for the reduced wild-type protein at pH 4. Solid black lines are experimental data and dashed red lines are simulations using parameters listed in Table 1. The lower inset shows the XANES region of the spectrum
Met to His, Cys, and Asp variants
To probe the role of the essential Met residue at position 471 in TBM, variants with His, Cys, or Asp replacing the Met were created. Figure 4 compares the spectra of all three mutants with those of the wild-type protein for the oxidized proteins. Remarkably, the agreement among all four experimental spectra is closer than that between the spectrum of the wild type and its best simulation. Therefore, we conclude that there is no difference in coordination between the wild type and the M471X variants, proving conclusively that residue 471 does not coordinate the CuM(II) center of the oxidized protein in a manner detectable by EXAFS spectroscopy. The spectra of the reduced variants differ significantly from the spectrum of the wild-type reduced protein (Fig. 5, top), but are all similar to each other (Fig. 5, bottom), suggesting that replacement of the M-site residue by His, Cys, or Asp produces a species with similar coordinate structure. All three reduced variants can be well fit by an average of two His residues at each copper with Cu–N(His) distances of 1.91 ± 0.01 Å (Fig. 6, Table 1). As expected, the His and Asp variants show no sign of Cu–S interactions. However, since the Cys variant exhibits small differences from His and Asp in the k = 7–9 Å−1 energy regime, we tested whether these arose from a Cu–S contribution of low shell occupancy, perhaps arising from a small population of enzyme molecules in a “Cys-on” conformation. When the Cu–S Debye–Waller factor was fixed at 0.005 Ǻ2, a value typical of Cu(I)–thiolate bonds, the simulations could tolerate 14% of a Cu–S conformation, with R Cu–S = 2.28 Ǻ. However, the goodness of fit was not improved in these simulations, and the inclusion of the Cu–S wave did not account for the small differences in EXAFS between k = 7 Å−1 and k = 9 Å−1. Therefore, the major species present in all three variants contains only Cu–His coordination, but we cannot exclude the possibility of a minor component in M471C having coordinated Cys, which might possibly account for the observed activity of this variant. Figure 6 compares experimental and simulated EXAFS spectra for the His and Cys variants. Simulation using a two-shell imidazole model resulted in one group of Cu–His interactions at a short distance of approximately 1.84 Å and a second group at a longer distance of approximately 1.95 Å. The separation of 0.11 Å is below the limit of resolution of the experiment given by ΔR = π/2Δk equal to 0.15 Å. These distances suggest one two-coordinate and one three-coordinate site, respectively.

Comparison of the experimental EXAFS (top) and phase-corrected Fourier transform data (bottom) for oxidized WT tyramine β-monooxygenase and its M-site Met to His, Asp, and Cys variants. WT black, M471H blue, M471C red, M471D green

Top: Comparison of experimental phase-corrected Fourier transforms and EXAFS (inset) of ascorbate-reduced WT (black trace) and the M471 to His variant (red trace) of tyramine β-monooxygenase. Bottom: Comparison of the experimental EXAFS data of the Met to His (blue), Met to Cys (red), and Met to Asp (green) variants

Phase-corrected Fourier transforms and EXAFS (upper insets) for reduced M471 to His (top) and reduced M471 to Cys (bottom) variants of tyramine β-monooxygenase. Solid black lines are experimental data and dashed red lines are simulations using parameters listed in Table 1. The lower insets show the XANES region of the spectrum
Absorption edges
XANES spectra for wild-type TBM and the M471 variants are shown in Fig. 7 (top). Wild type oxidized and reduced spectra are compared in Fig. 7 (top). These are typical for this class of copper monooxygenases, with the oxidized absorption edge shifted about 7 eV relative to the reduced protein with a low-intensity 1s → 3d transition at 8,979 eV, and weak unresolved features on the rising edge at 8,983.5 and 8,988 eV. The reduced protein exhibits a resolved feature at 8,983.4 which can be assigned as the 1s → 4p transition of Cu(I) in a three-coordinate environment [37, 39, 40]. All of these XANES features resemble those of PHM [23] and DBM [10], and confirm the similarity in coordinate structure of the copper centers of this class of copper monooxygenases.

Comparison of XANES of the copper centers of tyramine β-monooxygenase. Top: Oxidized versus reduced wild-type proteins. Bottom: Reduced proteins: WT black, Met to His blue, Met to Cys red, Met to Asp green
Figure 7 (bottom) compares XANES spectra for the reduced wild-type protein and the three M471 mutant proteins. The M471 variants also show the 1s → 4p transition but it is more intense and shifted slightly to higher energy (8,984.0 eV). Of particular interest is the almost exact coincidence of the XANES spectra for all three M471 variants, reinforcing the conclusion from EXAFS analysis that they all have very similar coordination at both copper centers. The increased intensity indicates that the average coordination number has decreased, and is consistent with the observed decrease in the Cu–N(His) distance, which drops from 1.94 Å in the reduced wild type to 1.91 Å in the mutant proteins. In general, Cu(I)–N(His) distances correlate with the coordination number, with the distance decreasing as the coordination number drops. The decrease in coordination can be explained by assuming that residue 471 no longer coordinates at the M-site, and that the latter is coordinated by just two His residues. Alternatively, the loss of M471 may also induce changes at the H-site as recently observed in the crystal structure of the M314I mutant of PHM [25].
Discussion
TBM shares structural features with PHM and DHM. Sequence alignment (Fig. 8) shows that all the residues known to be involved in copper binding in PHM are conserved in TBM, and suggests that TBM likewise contains a pair of mononuclear copper centers bound by H247, H248, and H317 (H-site) and H396, H398, and M471 (M-site). However, kinetic studies with TBM have revealed several important differences between the mechanism of the insect enzyme and the established mechanism of the mammalian homologues, with respect to the interaction of the enzymes with substrate and ascorbate [29]. We used XAS to investigate the coordination environment of the copper centers of TBM with the purpose of understanding the structural origins of these mechanistic differences. Data on the oxidized and reduced forms of the wild-type protein bear a close resemblance to those from PHM establishing a homology with respect to copper binding. Thus, we conclude that in oxidized TBM Cu(II) is bound by three His residues and a water molecule at the H-site, and by two His residues and two solvent molecules at the M-site. On reduction, the solvent coordination is lost, and M471 moves closer to CuM to form a three-coordinate center comprising H396, H398, and M471. Although the EXAFS data are best fit to two His residues at each copper site, the similarity of the spectra to previous data on reduced wild-type PHM and DBM, and the additional crystallographic information available for PHM, supports a three-coordinate CuH site, with one weakly bound His residue undetectable by EXAFS. Thus, loss of solvent at the H-site appears to result in a three-coordinate Cu(I) center in the reduced form of wild-type TBM, bound by H247, H248, and H317. Thus, mechanistic differences among the three enzymes do not appear to result from an altered copper coordination environment in TBM.

Sequence alignment using Clustal W 2.0.10 of members of the family of mononuclear monooxygenases. Sequences for peptidylglycine monooxygenase (PHM) and dopamine β-monooxygenase (DBM) are from rat, for tyramine β-monooxygenase (TBM) are from Drosophila melanogaster, and for monooxygenase X (MOX) are from human. Metal-binding residues and conserved Met residues are shown in red
The pH dependence of the EXAFS spectrum of reduced wild-type TBM shows an increase in intensity of the Cu–S interaction similar to that reported previously for PHM but with important differences [24]. Although the pK a for the pH transition was not determined, it is likely that a similar Met-on to Met-off transition controls the catalytic activity of TBM at low pH. In PHM, the Met-off form was shown to be catalytically active, whereas the Met-on form was inactive, and it was proposed that Met-off represented the weakly bound, fluxional CuM–S(Met) form fully described by EXAFS and crystallography. It was proposed [24] that Met fluxionality provided the necessary protein dynamics to bring the enzyme into the transient configuration critical for hydrogen tunneling. The identity of the Met-on form is less clear, but two possibilities exist: (1) a rigid, strongly bound CuM–S(Met) lacking in some essential fluxional component necessary for catalysis, or (2) a Cu–S interaction formed between either a copper center and a different sulfur-containing residue or, alternatively, reaction of one of the copper centers with an exogenous chloride ion. The fact that TBM low-pH data fit better to one Cu–S/Cl per TBM with a longer bond length yet smaller Debye–Waller factor is suggestive of the latter conclusion. All of the ten Cys residues in PHM are present as disulfides and it is therefore unlikely that additional Cu–S coordination involves a Cu(I)–Cys(thiolate) interaction. However, the strong preference of Cu(I) centers for Met coordination, for example in the periplasmic copper binding proteins CusF [38], CusB [41], pcoC [42], and CopK [43], indicates that a Met residue is a likely candidate. PHM and TBM share only two conserved Met residues, the catalytic-site M314/M471 residue and M109/M249, which is contiguous to the H-site copper-binding His residues, but is rotated away from the copper on the opposite side of the β-sheet. If the Met-on form represents copper coordination by a different Met residue, then CuH coordination to M109/M249 seems a likely option. This would require a significant conformational change induced by protonation of an as yet unassigned residue. The resultant loss of activity could be the result of a change in reduction potential of the H-site expected if a Met residue replaces a His residue. Alternatively, the reorientation of β-structure required to bring M109/M249 into coordinating distance of the CuH center would most likely disrupt the hydrogen bond between H108 and E170 (numbering refers to PHM) which has been proposed as part of the putative substrate-mediated electron transfer pathway between the two copper centers [15].
The possibility that the additional interaction is due to binding of exogenous chloride at low pH cannot be excluded on the basis of EXAFS analysis, but seems less likely from a chemical perspective. There is no reason why chloride binding should be pH-dependent as HCl is a strong acid. Additionally, similar chemistry has recently been observed with PHM in samples which do not contain chloride ion [24]. Therefore, although we cannot exclude chloride, coordination by M249 seems the most likely option.
The unique coordination of the copper centers has been shown in PHM and DBM to induce novel monooxygenase chemistry. In these sister proteins, catalysis begins by coordination of an oxygen molecule at the reduced mononuclear CuM center as observed crystallographically in a precatalytic complex with a slow substrate [16]. The O–O bond length in this complex is consistent with a Cu(II)–superoxo species, and dovetails with other biochemical [17, 19] and theoretical [3, 20] studies that implicate Cu(II)–O ·−2 as the reactive oxygen intermediate. The electrophilic nature of such an intermediate is expected to lead to efficient hydrogen-atom abstraction from an α-carbon of peptide substrate [44] or the benzylic position of phenethylamines, and calculations suggest a preference for side-on superoxide coordination [3, 20]. However, among the growing number of mononuclear Cu(II)–superoxo model complexes [26], only two are reactive toward hydrocarbon substrates [45, 46], and these exhibit end-on superoxo coordination. A more common reactivity of mononuclear Cu(II)–superoxo species is dimerization with another molecule of the Cu(I) parent complex to generate the dinuclear Cu(II)–peroxo complex. The large spatial separation of the copper centers in PHM and by analogy TBM prevents this chemistry, and thus allows the potent electrophilic reactivity of the mononuclear Cu(II)–superoxo species to be fully expressed in the form of hydrogen-atom abstraction from the substrate to form a [CuM(II)-OOH] (peroxo) intermediate. Subsequent steps in the reaction pathway are less clear and alternative mechanisms have been proposed that involve long-range electron transfer from CuH either before [17] or after [20] the transfer of an OH group to the substrate radical.
An unresolved issue relating to this mechanism is the role of the M-site Met residue in stimulating catalysis. As stated earlier, the flexibility of the Met ligand has been invoked as a necessary element for catalysis. In addition, the Met ligand is believed to influence the Cu(II)/(I) redox equilibrium at the CuM site [17] and thus affect the extent of dioxygen activation. A preference for formation of an initial Cu(I)–O2 versus a Cu(II)–superoxide species is thought to stem from the effect of sulfur ligation. This equilibrium shift in favor of an unactivated bound dioxygen species would greatly reduce uncoupling and the formation of harmful reactive oxygen species, prior to C–H activation. The alternative residues Asp, His, and Cys, in place of the Met ligand at CuM, were thus chosen to further examine the significance of this residue.
Mutation of M471 in TBM to His, Asp, or Cys resulted in a large decrease in activity, with only the M471C variant displaying measurable activity, albeit coincident with enzyme inactivation [9]. In the case of PHM, the M314X variants had no detectable activity in assays of spent media from Chinese hamster ovary cell overexpression [21, 22]. One of the goals of the present work was to provide a structural basis for the reduction/loss in catalytic activity by examining the EXAFS-derived structures of the M471X (X is His, Cys, Asp) variants. Results on the oxidized protein establish that the Met ligand does not bind strongly to the Cu(II)M center, as no Cu–S interaction is observed. This result is similar to those for PHM and DBM, where no Cu–S EXAFS interaction is seen in the Cu(II) form [10, 23], owing to a long axial Cu–S(Met) interaction [13], and leads to the prediction that His and Asp would likewise bind axially and be undetectable by EXAFS, as is observed experimentally. The Cys variant might have been expected to show some properties of type 1 copper centers due to the N2S coordination, but it too appears to produce no perturbation on the wild-type spectrum with no observable thiolate-to-Cu(II) charge transfer. The EXAFS results together with earlier EPR data [9] therefore suggest that the oxidized CuM site accommodates all four side chains without significant structural rearrangement.
In contrast, the reduced forms of wild-type and variant TBM show significant differences. At pHs at or above the activity maximum (pH 5.5–6), reduced wild-type TBM exhibits a Cu–S interaction equivalent to 0.5 Cu–S at 2.25 Å with a Debye–Waller factor (2σ 2 = 0.015 Å2) which is high for a strongly coordinated ligand, and may suggest some fluxional behavior involving either multiple Met conformations (as is seen, for example, in the periplasmic chaperone CusF [38]), or alternatively rapid coordination and dissociation of the thioether moiety. Such behavior has been observed in Cu(I)N X S(thioether) models of the CuM site including pendant thioether containing β-diketiminate Cu(I) complexes which show temperature-dependent changes in the thioether-bonded methylene proton NMR resonances [28], and in TMPA-derived (TMPA = tris(2-pyridylmethyl)amine) N3S(thioether) models where the thioether arm is easily displaced by CO [47]. Cu–S(Met) fluxionality may be an important feature of the M active site, either to provide facile conversion to axially coordinated Cu(II)–S(Met) upon oxygen binding and formation of the Cu(II)–superoxo intermediate [13], or to provide kinematic coupling to a normal mode critical to hydrogen tunneling during the hydrogen-atom abstraction step [1, 24, 48].
Comparison of the EXAFS data for the His, Asp, and Cys variants with EXAFS data for reduced wild-type TBM shows differences, but analysis leads to the conclusion that none of these substitutions lead to detectable coordination to copper. The presence of both CuH and CuM makes it difficult to isolate the structural changes at the M-site, and the data give only the average change at both coppers. However, since the H-site locus is unchanged in these variants, it is reasonable to assume that the observed structural changes derive mainly from the M-site substitutions. Analysis of the EXAFS data gives a similar structure for all three variants, with loss of the Cu–S component, and decrease in the residual two Cu–N(His) bond lengths. In parallel, the XANES data shows an increase in intensity of the 8,983-eV edge feature. These changes are consistent with a decrease in coordination number and suggest a simple interpretation that the M-site becomes two-coordinate. Although coordination of oxygen-donor groups, such as Asp, is less common among copper proteins, it is surprising that neither His nor Cys seems capable of significant binding to CuM. The fluxional Cu(I)–Met interaction, and its movement by some 0.5 Å on copper oxidation indicates the absence of steric restrictions in the site, and modeling confirms this conclusion (Fig. 1c). In other systems, Met to His substitution appears quite facile, for example, the M121H variant of Alcaligenes denitrificans azurin, where despite the “rack-induced” rigidity of the cupredoxin fold, H121 moves closer to the copper and becomes a fourth strongly bound ligand [49]. Likewise, recent work from one of the authors’ laboratories (N.J.B.) has demonstrated Met coordination in the H135 to Met substitutions of Bacillus subtilis Sco (unpublished).
The crystal structure of the PHM M314I variant suggests one possible reason for the failure of the alternative His or Cys residues to coordinate [25]. Here the CuM site in oxidized M314I PHM accommodates the mutation by replacing M314S γ with a water molecule, and by shifting the positions of the other coordinating residues (H242, H244, and a water molecule), to form a distorted tetrahedron. I314 is rotated away from the copper in a different conformation. Interestingly the PHM M314I structure also shows significant perturbation at the H-site. H107 and H172 are now linearly coordinated with H108 dissociated. The M471X substitutions in TBM may favor this altered conformation. Therefore, one hypothesis for the essential role of Met at the M-site is that it uniquely stabilizes the protein in its catalytic conformation in addition to, or instead of, electronic tuning of the reactivity of the Cu(II)–superoxo [17] and/or the Cu(II)–oxyl intermediates [20] as previously proposed.
This does not, however, explain the singular activity of the M471C TBM mutant. The lack of sulfur-atom coordination by the Cys residue to CuM(I) does not preclude substrate turnover. The observed activity of the M471C mutant may arise from a small population of enzyme in a Cys-bound conformation, which is not inconsistent with the EXAFS analysis. Low occupancy of the Cys-bound form could also provide an explanation for the inactivation of M471C TBM during the reaction with substrate. According to the current hypothesis, the fluxional dynamics of the Cu–Met bond prevent uncoupling of dioxygen and C–H activation during the catalytic cycle. Since a fully liganded Cys may be expected to modulate the chemistry at the CuM site as efficiently as the Met ligand, the inactivation of M471C via oxidative damage (R.L. Osborne and J.P. Klinman, unpublished data) may be a direct consequence of its poor coordination to the reduced metal. Further work is needed to evaluate these aspects.
Abbreviations
- DBM:
-
Dopamine β-monooxygenase
- EXAFS:
-
Extended X-ray absorption fine structure
- FT:
-
Fourier transform
- PHM:
-
Peptidylglycine monooxygenase
- S2:
-
Schneider 2
- TBM:
-
Tyramine β-monooxygenase
- Tris:
-
Tris(hydroxymethyl)aminomethane
- XANES:
-
X-ray absorption near edge structure
- XAS:
-
X-ray absorption spectroscopy
References
Klinman JP (2006) J Biol Chem 281:3013–3016
Prigge ST, Mains RE, Eipper BA, Amzel LM (2000) Cell Mol Life Sci 57:1236–1259
Chen P, Solomon EI (2004) Proc Natl Acad Sci USA 101:13105–13110
Gray EE, Small SN, McGuirl MA (2006) Protein Expr Purif 47:162–170
Xin X, Mains RE, Eipper BA (2004) J Biol Chem 279:48159–48167
Roeder T (2005) Annu Rev Entomol 50:447–477
Lehman HK, Schulz DJ, Barron AB, Wraight L, Hardison C, Whitney S, Takeuchi H, Paul RK, Robinson GE (2006) J Exp Biol 209:2774–2784
Monastirioti M (2003) Dev Biol 264:38–49
Hess CR, Wu Z, Ng A, Gray EE, McGuirl MA, Klinman JP (2008) J Am Chem Soc 130:11939–11944
Blackburn NJ, Hasnain SS, Pettingill TM, Strange RW (1991) J Biol Chem 266:23120–23127
Pettingill TM, Strange RW, Blackburn NJ (1991) J Biol Chem 266:16996–17003
Boswell JS, Reedy BJ, Kulathila R, Merkler DJ, Blackburn NJ (1996) Biochemistry 35:12241–12250
Chen P, Bell J, Eipper BA, Solomon EI (2004) Biochemistry 43:5735–5747
Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM (1997) Science 278:1300–1305
Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM (1999) Nat Struct Biol 6:976–983
Prigge ST, Eipper BA, Mains RE, Amzel M (2004) Science 304:864–867
Evans JP, Ahn K, Klinman JP (2003) J Biol Chem 278:49691–49698
Evans JP, Blackburn NJ, Klinman JP (2006) Biochemistry 45:15419–15429
Bauman AT, Yukl ET, Alkevich K, McCormack AL, Blackburn NJ (2006) J Biol Chem 281:4190–4198
Chen P, Solomon EI (2004) J Am Chem Soc 126:4991–5000
Eipper BA, Quon ASW, Mains RE, Boswell JS, Blackburn NJ (1995) Biochemistry 34:2857–2865
Kolhekar AS, Keutman HT, Mains RE, Quon ASW, Eipper BA (1997) Biochemistry 36:10901–10909
Blackburn NJ, Rhames FC, Ralle M, Jaron S (2000) J Biol Inorg Chem 5:341–353
Bauman AT, Jaron S, Yukl ET, Burchfiel JR, Blackburn N (2006) Biochemistry 45:11140–11150
Siebert X, Eipper BA, Mains RE, Prigge ST, Blackburn NJ, Amzel LM (2005) Biophys J 89:3312–3319
Itoh S (2006) Curr Opin Chem Biol 10:115–122
Aboelella NW, Kryatov SV, Gherman BF, Brennessel WW, Young VG Jr, Sarangi R, Rybak-Akimova EV, Hodgson KO, Hedman B, Solomon EI, Cramer CJ, Tolman WB (2004) J Am Chem Soc 126:16896–16911
Aboelella NW, Gherman BF, Hill LM, York JT, Holm N, Young VG Jr, Cramer CJ, Tolman WB (2006) J Am Chem Soc 128:3445–3458
Hess CR, McGuirl MM, Klinman JP (2008) J Biol Chem 283:3042–3049
George GN (1990) Exafspak. Stanford Synchrotron Radiation Laboratory
Binsted N, Gurman SJ, Campbell JW (1998) Excurve 9.2. Daresbury Laboratory
Gurman SJ, Binsted N, Ross I (1984) J Phys C 17:143–151
Gurman SJ, Binsted N, Ross I (1986) J Phys C 19:1845–1861
Binsted N, Hasnain SS (1996) J Synchrotron Radiat 3:185–196
Sanyal I, Karlin KD, Strange RW, Blackburn NJ (1993) J Am Chem Soc 115:11259–11270
Himes RA, Park GY, Siluvai GS, Blackburn NJ, Karlin KD (2008) Angew Chem Int Ed 47:9084–9087
Himes RA, Park YG, Barry AN, Blackburn NJ, Karlin KD (2007) J Am Chem Soc 129:5352–5353
Loftin IR, Franke S, Blackburn NJ, McEvoy MM (2007) Protein Sci 16:2287–2293
Kau LS, Spira-Solomon D, Penner-Hahn JE, Hodgson KO, Solomon EI (1987) J Am Chem Soc 109:6433–6442
Pickering IJ, George GN, Dameron CT, Kurz B, Winge DR, Dance IG (1993) J Am Chem Soc 115:9498–9505
Bagai I, Liu W, Rensing C, Blackburn NJ, McEvoy MM (2007) J Biol Chem 282:35695–35702
Peariso K, Huffman DL, Penner-Hahn JE, O’Halloran TV (2003) J Am Chem Soc 125:342–343
Sarret G, Favier A, Coves J, Hazemann JL, Mergeay M, Bersch B (2010) J Am Chem Soc 132:3770–3777
Hatcher LQ, Karlin KD (2004) J Biol Inorg Chem 9:669–683
Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S (2009) J Am Chem Soc 131:2788–2789
Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD (2007) J Am Chem Soc 129:264–265
Lee DH, Hatcher LQ, Vance MA, Sarangi R, Milligan AE, Sarjeant AA, Incarvito CD, Rheingold AL, Hodgson KO, Hedman B, Solomon EI, Karlin KD (2007) Inorg Chem 46:6056–6068
Klinman JP (2006) Biochim Biophys Acta 1757:981–987
Messerschmidt A, Prade L, Kroes SJ, Sanders-Loehr J, Huber R, Canters GW (1998) Proc Natl Acad USA 95:3443–3448
Acknowledgments
We thank Andrew Bauman for help with collection of XAS data. We gratefully acknowledge the use of facilities at the Stanford Synchrotron Radiation Lightsource, which is supported by the National Institutes of Health Biomedical Research and Technology Program Division of Research Resources, and by the US Department of Energy Office of Biological and Environmental Research. The work was supported by grants from the National Institutes of Health (R01 NS027583 to N.J.B, R01 GM0257651 to J.P.K., and GM067351 to C.H.).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hess, C.R., Klinman, J.P. & Blackburn, N.J. The copper centers of tyramine β-monooxygenase and its catalytic-site methionine variants: an X-ray absorption study. J Biol Inorg Chem 15, 1195–1207 (2010). https://doi.org/10.1007/s00775-010-0677-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00775-010-0677-3