
Abstract
Plasmid DNA vaccines are considered alternatives to inactivated influenza virus vaccines to control influenza. Vaccination with a hemagglutinin (HA)-, HA ectodomain (HAe)-, or HA subunit 1 (HA1)-based vaccine can stimulate protective immunity in animals. The aim of this study was to compare their capacity to induce an antibody response and protection against influenza virus infection in mice after DNA vaccination. We constructed three expression vectors encoding full-length HA, HAe, or HA1 of the A/California/07/2009 influenza A virus and designed three animal experiments: (i) BALB/c mice were immunized twice with 30 μg of the HA, HAe, or HA1 DNA vaccine with high-voltage electroporation (100 V), and 3 weeks after boosting, they were challenged with a lethal dose of virus. (ii) Immunization and challenge were as in experiment i, but with low-voltage electroporation (10 V). (iii) Mice were immunized once with 50 μg of DNA and challenged 1 week later. The immunogenic effects of the three DNA vaccines were evaluated in terms of antibody titer, survival rate, bodyweight change, and lung viral titer. In all three experiments, both HA and HAe induced higher antibody and neutralization titers than HA1. Following challenge with a lethal mouse-adapted homologous virus, both HA and HAe reduced the viral titers in lung washes or offered better protection from weight loss than HA1 in experiments ii and iii. Thus, HA1 induces a lower immune response than HA or HAe when used as a DNA vaccination. Our data should be valuable in choosing the optimal candidate vaccine when faced with the threat of pandemic influenza.
Similar content being viewed by others

Introduction
In 2009, a flu pandemic presented a global threat to public health. It began in Mexico and was caused by a new strain of the H1N1 influenza virus [1]. The pandemic H1N1 2009 virus (H1N1pdm09) spread rapidly around the world after the initial outbreak. By the time the World Health Organization (WHO) [2] had declared a pandemic, 74 countries and territories had reported laboratory-confirmed infections. By the end of 2009, this new virus had caused 10,000 deaths. H1N1pdm09 is a novel influenza virus, with a genome that is a combination of North American and Eurasian swine influenza virus lineages [3–5]. Although no donor viruses were isolated from swine, this novel virus had possibly been circulating for some time in swine before crossing the species barrier to infect humans [4]. Like other influenza viruses, the person-to-person transmission of H1N1pdm09 occurs via respiratory droplets [6], and H1N1pdm09 appears to spread among people with greater ease than the seasonal flu virus [7]. In vivo experiments have also shown that this virus has a greater capacity to proliferate than seasonal influenza viruses [8–10]. Because H1N1pdm09 shares low levels of genetic and antigenic similarity with seasonal influenza viruses [3], vaccines against these seasonal viruses produce relatively low immunity to H1N1pdm09 [11, 12].
Vaccination is the most effective strategy for combating an influenza pandemic. In 2009, monovalent inactivated vaccines were produced as quickly as possible once the outbreak was identified. Clinical results showed that adults immunized with an inactivated vaccine displayed greater than 90 % seroconversion [13, 14]. In China, a vaccine production program was launched as early as June 2009, and the inactivated vaccine was available several months later. Clinical results showed that the inactivated vaccine induced satisfactory results in all age groups [15, 16]. Although the inactivated 2009 H1N1 influenza vaccines have proved effective in eliciting neutralizing antibodies against the virus in clinical trials, other candidate vaccines against H1N1pdm09 must be developed. Plasmid DNA vaccines have been considered alternatives to inactivated influenza virus vaccines for controlling influenza. Many studies have shown that DNA vaccines can protect animals against influenza A viral infections [17–19], and in our previous studies, we have demonstrated that DNA vaccination in animal models effectively prevented influenza virus infection [18, 20, 21].
The viral hemagglutinin (HA) protein is a homotrimer. Each of its single-chain monomers is initially synthesized as a precursor polypeptide, HA0, which is then cleaved by host proteases into two subunits, HA1 and HA2 [22, 23]. The HA protein of the influenza A virus is the major antigen that elicits protective immune response. Antibodies directed against this surface glycoprotein provide protection by blocking viral attachment and entry [24]. HA is the main component of inactivated vaccines directed against seasonal influenza viruses and also acts as the major antigen for most DNA vaccines [21, 25], subunit vaccines [26, 27], and viral-vector-based vaccines [28–30]. The ectodomain of HA (HAe) and HA1, two forms of truncated HA, has also been used as a vaccine to immunize animals. Previous studies have demonstrated that a vaccine based on HAe, an HA1 region of the influenza A virus, induces neutralizing activity and protection against influenza viral challenge [31–36], suggesting that HAe and HA1 of the HA protein play significant roles in the immune response to viral infection and are attractive targets for vaccine development. However, no study has compared the immunogenicity of HA, HAe, and HA1 in mice after DNA vaccination.
In the work reported here, plasmids expressing the full-length HA protein or truncated forms of HA (HAe and HA1) were constructed. We determined the cellular localization of the antigens and compared their ability to induce an antibody response and protection against influenza virus infection in mice after DNA vaccination.
Materials and methods
Virus preparation
Strain NYMC X-179A [A/reassortant/NYMC X-179A (California/07/2009 x NYMC X-157)(H1N1)] was generated by New York Medical College and supplied by the Centers for Disease Control and Prevention (USA). This strain is a pandemic vaccine strain recommended for use in vaccine development [14]. The seed virus was grown in embryonated eggs with the fifty percent tissue culture infection dose (TCID50) determined in Madin-Darby canine kidney (MDCK) cells according to the Reed-Muench method [37].
Other influenza virus strains [A/PR/8/34 (H1N1), A/New Caledonia/20/1999 (H1N1), A/Chicken/Henan/12/2004 (H5N1) and A/Chicken/Jiangsu/7/2002 (H9N2)] were also used. Viruses were propagated in the allantoic cavities of 10-day-old embryonated chicken eggs at 37 °C for 48 h and stored at −80 °C until required. H5N1 virus was handled in a biosafety level 3 laboratory.
Mouse-adapted virus
All animal experiments were conducted in accordance with ethical procedures and policies approved by the Wuhan Institute of Virology’s Institutional Animal Care and Use Committee. Prior to animal infection for evaluation of the protective efficacy of DNA immunization, a mouse-adapted virus was prepared. Strain NYMC X-179A (H1N1) is a pandemic vaccine strain recommended for use in vaccine development [14]. The seed virus was almost completely avirulent for mice. To enhance the virulence of NYMC X-179A, we produced a mouse-adapted strain by lung-to-lung passage in mice. Three BALB/c mice were anesthetized, and each was inoculated with 50 μl of the NYMC X-179A viral suspension (104 TCID50) by intranasal drip. At 3-5 days post-inoculation, mice were sacrificed and their trachea and lungs removed. Tissues were washed three times in a total volume of 2 ml of phosphate-buffered saline (PBS) containing 0.1 % bovine serum albumin (BSA). The bronchoalveolar washes were collected and used to infect the next batch of mice after removing the cellular debris by centrifugation. The lung-to-lung passage tests were repeated until the virus was lethal for mice. After twenty serial passages, mice developed clinical symptoms, including hunched posture, weakness, weight loss, and ruffled fur, and the viruses exhibited high virulence in mice. We then plaque purified two of the mouse-passaged viruses. The final adapted virus was harvested, aliquoted, and stored at −80 °C. The 50 % mouse lethal dose (LD50) of each stock was determined using the Reed-Muench method [37].
Plasmid DNAs
The full-length HA gene from the 2009 H1N1 strain A/California/04/2009 virus was ligated into the pUC/HA vector (Shanghai Sangon Biological Engineering Technology & Services Co., Ltd.). Plasmid pCAGGSP7/HA was constructed by amplifying full-length HA from pUC/HA using primers HA-F (5′-AAA CTC GAG AGC AAA AGC AGG GGA AAA TAA A-3′) and HA-R (5′-AAT CCC GGG AGT AGA AAC AAG GGT GTT TTT T-3′) and then ligating into the expression vector pCAGGSP7 between the XhoI and SmaI restriction endonuclease sites.
The HAe fragment was amplified from pCAGGSP7/HA using primers HA-F and HAe-R (5′-AAT CCC GGG TCA CTG GTA AAT CCT TGT TGA TTC C-3′). The HA1 fragment was amplified from pCAGGSP7/HA using primers HA-F and HA1-R (5′-AAT CCC GGG TCA TCT AGA TTG AAT AGA CGG GAT A-3′). HAe and HA1 were ligated into pCAGGSP7 between the XhoI and SmaI restriction endonuclease recognition sites to yield pCAGGSP7/HAe and pCAGGSP7/HA1, respectively. The nucleotide sequences of HA, HAe and HA1 were confirmed by sequencing. The expression levels of the proteins encoded by the HA, HAe and HA1 DNA sequences were confirmed in 293T human embryonic kidney cells as described previously [38]. Plasmids were propagated in Escherichia coli DH5α and purified using a QIAGEN-tip 500 plasmid purification kit (QIAGEN, Germany).
Indirect immunofluorescence
COS-7 cells were co-transfected with 2 μg of pCAGGSP7/HA, pCAGGSP7/HAe or pCAGGSP7/HA1 using Lipofectamine 2000 (Invitrogen) in serum-free medium. After incubation for 5 h, the medium was replaced with fresh medium containing 10 % fetal bovine serum. At 24 h post-transfection, cells were washed with PBS, fixed with 4 % paraformaldehyde (pH 7.4) for 30 min, permeabilized with 0.2 % Triton X-100 in PBS for 30 min, and stained with Hoechst 33258 for 30 min. Indirect immunofluorescent staining was performed with an HA (NYMC X-179A)-specific rabbit antiserum and FITC-labeled goat anti-rabbit antisera. Fluorescence imaging was performed with a TCS-SP2 confocal microscope (Leica, Germany) equipped with a cooled CCD camera. All measurements were obtained with a 100 × oil immersion objective (NA 1.32) and 2× zoom.
DNA immunization by electroporation
In vivo electroporation was carried out according to the method described by Aihara and Miyazaki [39]. Adult female BALB/c mice (6–8-weeks old) were immunized with plasmid DNA dissolved in 30 μl of Tris–EDTA buffer. After injection in the right quadriceps muscle, a pair of electrode needles 5 mm apart was inserted into the muscle to cover the DNA injection sites, and electric pulses were delivered using an electric pulse generator (ECM830; BTX, San Diego, CA).
Serum antibody assays
Enzyme-linked immunosorbent assays (ELISAs) were performed in 96-well plates (Costar, Cambridge, MA). Reagents used in the assays included: inactivated vaccine against NYMC X-179A diluted to 10 μg/ml (Shanghai Institute of Biological Products, Shanghai, China), serial two-fold dilutions of sera from each group of immunized or unimmunized mice; goat anti-mouse IgG (α-chain specific) (Southern Biotechnology Associates, Inc., USA) conjugated to biotin, streptavidin conjugated to alkaline phosphatase (Southern Biotechnology Associates), and p-nitrophenyl-phosphate. The amount of chromogen produced was measured based on the absorbance measured at 410 and 630 nm using an ELISA reader (GENios, Tecan).
HAI assay
The hemagglutinin inhibition (HAI) assay was performed as described previously [40]. Briefly, the sera were treated with receptor-destroying enzyme (Denke-Seiken, Japan) and inactivated at 56 °C for 30 min, followed by incubation with chicken erythrocytes to adsorb nonspecific agglutinins. The sera from each group of immunized or unimmunized mice were serially diluted twofold with PBS in a 96-well polystyrene microtiter plate, 25 μl in each well. A portion of 25 μl of virus suspension containing 4 hemagglutinin units (HAU) was added to each well. After incubation of the plate at room temperature for 1 h, 50 μl of 0.5 % (v/v) chicken red blood cells was added to each well, and the plate was incubated at room temperature for 30 minutes. The HAI titers were determined as the highest serum dilution that completely inhibited hemagglutination.
Microneutralization assay
Titers of neutralizing antibodies (NAbs) were determined as described previously [41]. The serum treated with receptor-destroying enzyme (RDE) was diluted from 1:20 to 1:2560 in twofold serial dilutions in culture medium (DMEM containing 100 U/ml penicillin G, 100 μg/ml streptomycin and 0.5 μg/ml TPCK-treated trypsin). Serum dilution solutions were mixed with culture medium containing 100 TCID50 of NYMC X-179A at room temperature for 1 h. The virus-serum mix was then transferred to MDCK cells. Culture medium (200 μl) was added, and the plates were incubated for 72 h. Endpoints were determined by hemagglutination titer.
Lethal challenge
Mice were anesthetized and challenged with the mouse-adapted virus strain at 10 × LD50 by intranasal administration in 50 μl of the viral suspension. This infection caused rapid and widespread viral replication in the lung and death of the unimmunized mice within 7 days.
Specimens
Mice were anaesthetized with chloroform and then bled from the heart with a syringe. Serum was collected from the blood and used in antibody assays. After bleeding, a ventral incision was made along the median line from the xiphoid process to the point of the chin. The trachea and lungs were removed and washed twice by injecting 2 ml of PBS containing 0.1 % BSA. The bronchoalveolar wash was used for virus titration after removing cellular debris by centrifugation [42].
Virus titrations
The bronchoalveolar wash was serially diluted tenfold and used to inoculate MDCK cells, which were then incubated at 37 °C and tested for hemagglutination activity 3 days later. The virus titer of each specimen, expressed as the TCID50, was calculated by the Reed-Muench method [37]. The virus titer in each experimental group was represented by the mean ± the standard deviation (S.D.) of the virus titer per ml of specimens from all mice in each group.
Statistical analysis
Comparisons of experimental groups were evaluated by Student’s t-test where a P-value less than 0.05 were considered significant. For survival, probability was calculated using Fisher’s exact test, comparing the rate of survival in mice immunized with the viral protein-expressing DNA with those of the control groups.
Results
Plasmid DNA construction and expression in mammalian cells
DNA fragments encoding amino acid sequences 1–566, 1–531, and 1–344 of HA from influenza A virus strain A/California/07/2009 were individually cloned into the expression vector pCAGGSP7 to generate pCAGGSP7/HA, pCAGGSP7/HAe, and pCAGGSP7/HA1, respectively (Fig. 1a). To detect the localization of each protein encoded by the plasmids in mammalian cells, COS-7 cells were transfected with each plasmid and protein localization was determined by indirect immunofluorescence. As shown in Fig. 1b, fluorescent signals were detected in cells transfected with each kind of plasmid DNA. Most of the HA protein localized on the cell membrane, whereas HAe and HA1 mainly localized in the cytoplasm. This result shows that the loss of the transmembrane domains of HAe and HA1 hindered their attachment to the cell surface, as observed in previous studies [43, 44].

a Schematic diagram of DNA vaccine constructs. Details of the design and construction of each plasmid are given in the Materials and methods. b Immunofluorescent confocal microscopic images. The indicated cDNAs were expressed in COS-7 cells. Indirect immunofluorescent staining was performed with a specific antibody against hemagglutinin (HA; clone NYMC X-179A) raised in rabbits, followed by a secondary fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit antibody
Adaptation of NYMC X-179A in mice
To identify any amino acid substitutions introduced during viral passage in mice, the sequences of HA and neuraminidase (NA) from mouse-adapted influenza A virus strain NYMC X-179A were compared with those of the wild-type virus NYMC X-179A. These two virus clones have identical viral genomes, but we found three amino acid differences between the mouse-adapted and wild-type viruses, one in HA (K145E) and two in NA (Q25R, I210M) (data not shown). Some mutations, such as K119N, D131E, G155E, S183P, A198E, R221K, and D222G in HA1 of pandemic H1N1 influenza virus, have been reported to occur during viral adaptation in the mouse lung [45, 46]. However, the K145E mutation in HA identified in this study is unique. NA mutations Q25R and I210M have also never been investigated. To determine whether the amino acid changes observed in mouse-adapted NYMC X-179A occur in other field strains, we selected 1000 field strains (in 2009–2012, full-length sequences only) from the flu virus gene database (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html) and analyzed the mutations at the corresponding sites. Residue 145K is highly conserved among the 2009 H1N1 isolates, with only five K145R mutations and no K145E mutations observed. The mutation Q25R in the NA protein was present in two of the H1N1 strains examined (2/1000), whereas the other mutation, I210M, was not observed in any of the field strains (data not shown).
Antibody-inducing capacity and protective efficacy of HA, HAe, and HA1 DNA vaccines in mice after immunization with high-voltage electroporation
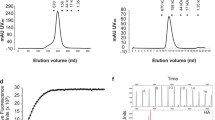
To compare the immune responses and protection against viral infection elicited by these three constructs, 6- to 8-week-old female BALB/c mice (n = 10 per group) were immunized with 30 μg of pCAGGSP7/HA, pCAGGSP7/HAe, or pCAGGSP7/HA1 with high-voltage electroporation (100 V). Nonimmunized mice were used as the control group. The animals were immunized twice after a 3-week interval. Serum was collected from the mice 3 weeks after boosting. Immunoglobulin G (IgG) titers and those of other antibodies were analyzed after the sera were treated with RDE. As shown in Fig. 2a, immunization with the HA, HAe, or HA1 DNA vaccine induced significant antibody responses compared with that in the control group (p < 0.01). Mice immunized with HA had significantly higher IgG titers than those immunized with HA1 (p = 0.006). The antibody titers of the HAe- and HA1-immunized mice did not differ significantly, although HAe induced slightly higher IgG titers than HA1. Consistent with the results of enzyme-linked immunosorbent assay (ELISA) studies, the antibodies induced by HA, HAe, and HA1 exhibited significantly higher hemagglutinin inhibition (HAI) and neutralization titers than those induced in the control group. The antibodies induced by the HA1 DNA vaccine also exhibited lower HAI and neutralization titers than those induced by HA or HAe (Fig. 2b, c), although these differences were not significant. We also analyzed the cross-neutralization titers of the antisera from mice immunized with HA, HAe, or HA1. Several strains of influenza virus, including A/PR/8/34 (H1N1), A/New Caledonia/20/1999 (seasonal H1N1), A/Chicken/Henan/12/2004 (H5N1), and A/Chicken/Jiangsu/7/2002 (H9N2), were selected for detection. No cross-neutralizing antibodies were detected against these viruses (data not shown).

BALB/c mice were immunized twice with 30 μg of pCAGGSP7/HA, pCAGGSP7/HAe, or pCAGGSP7/HA1 with high-voltage electroporation (100 V). Three weeks after boosting, the sera were collected for the titration of IgG, HAI, and NAbs. a Titers of IgG in the mouse sera after boosting. Anti-HA IgG titers are expressed as the highest serum dilution that yielded an optical density greater than twice the mean of similarly diluted negative control samples. The data shown are the mean antibody titers of the four mice in each group with standard errors (error bars). b HAI titers in vaccinated mice. HAI titers of the immune sera were determined as the capacity of the sera to inhibit virus hemagglutination of chicken red blood cells. Representative data are geometric means ± SD. c NAb titers in vaccinated mice. NAb titers are shown as the geometric mean for each group with SD (error bars). Three weeks after boosting, the mice were challenged with a lethal dose of mouse-adapted NYMC X-179A (10 × LD50). The survival rates, bodyweight changes, and lung viral titers were determined. d Percentage survival of mice infected with lethal virus. Mouse survival was monitored daily for 2 weeks. e Bodyweight loss. Bodyweight loss was observed for 14 days after infection. f Lung viral titers on day 3 post challenge. The mice were sacrificed 3 days after viral challenge. The tracheae and lungs were removed and washed twice by injection of 2 ml of PBS containing 0.1 % bovine serum albumin. The bronchoalveolar wash was used for virus titration after the cellular debris was removed by centrifugation
To determine the protective efficacy of the HA, HAe, and HA1 DNA vaccines against viral infection, mice were challenged with 10 × LD50 of the mouse-adapted virus 3 weeks after boosting. Three and 5 days after infection, almost no weight loss was observed in the mice immunized with the DNA vaccines, whereas the mice in the control group exhibited approximately 15 % and 25 % weight loss, respectively (Fig. 2e). All mice immunized with the DNA vaccines were still alive 2 weeks after infection (Fig. 2d), and very low viral titers were detected in their lungs (Fig. 2f). The mice in the control group died within 1 week, with high viral titers in their lungs. Thus, two successive immunizations of mice with the HA, HAe, or HA1 DNA vaccine with high-voltage electroporation induced a robust immune response and provided complete protection against lethal viral challenge, although HA1 induced a slightly lower antibody response than that induced by HA or HAe.
Comparison of HA, HAe, and HA1 DNA vaccination with low-voltage electroporation in mice
In a previous study, we demonstrated that electroporation following intramuscular injection markedly enhanced the efficacy of immunization and provided effective protection, even at a voltage of 10 V [47]. However, the antibody titers induced in mice gradually decreased as the voltage decreased [47]. To investigate whether immunization with the HA, HAe, or HA1 DNA vaccine with electroporation at a voltage of 10 V could induce a protective immune response against viral infection, female BALB/c mice (n = 10 per group) were immunized and challenged as described above, except with low-voltage electroporation (10 V). Compared with the experiment described above, apparent reductions in the IgG antibody, HAI, and neutralizing titers were observed with electroporation at 10 V. As shown in Fig. 3(a), mice immunized with HA and HAe displayed higher IgG titers than mice immunized with HA1. Consistent with the results of ELISAs, the antibodies induced by the HA1 DNA vaccine displayed lower HAI and neutralization titers than those induced with HA and HAe, although the differences were not significant (Fig. 3b, c).

BALB/c mice were immunized twice with 30 μg of pCAGGSP7/HA, pCAGGSP7/HAe, or pCAGGSP7/HA1 with low-voltage electroporation (10 V). Three weeks after boosting, the sera were collected for the titration of IgG, HAI, and NAbs. a Titers of IgG in mouse serum after boosting. b HAI titers in vaccinated mice. c NAb titers in vaccinated mice. Seven days after immunization, mice were challenged with a lethal dose of mouse-adapted NYMC X-179A (10 × LD50). Survival rates, bodyweight changes, and lung viral titers were determined. d Percentage survival of mice infected with lethal virus. e Bodyweight losses. f Lung viral titers on day 3 post-challenge
To determine whether this reduced immune response is associated with lower protective efficacy against viral infection, mice were challenged with 10 × LD50 of the mouse-adapted virus. All immunized mice survived for 2 weeks (Fig. 3d), although different levels of weight loss were observed. Three and 5 days after infection, weight losses of approximately 5 % and 3 %, respectively, were observed in the HA-immunized mice; 9 % and 8 %, respectively, in the HAe group; and 18 % and 22.5 %, respectively, in the HA1-immunized mice. The mice in the HA1 group showed a significantly greater loss in bodyweight than the mice in the HA and HAe groups (p < 0.01). This demonstrates that viral infection caused more-severe clinical signs of illness in the HA1-immunized mice. We then determined the viral loads in the lungs at 3 days postinfection (dpi). The results demonstrated that HA and HAe significantly reduced the viral loads in the lungs compared with the high viral loads of the control group (p < 0.05). Consistent with the weight loss results, high viral loads were detected in the lungs of the mice of the HA1 group. Thus, immunization with the HA or HAe DNA vaccine using low-voltage electroporation was more effective in inducing protective immunity against a lethal viral challenge than immunization with HA1.
HA1 induces less-efficient early protection than HA or HAe
In our previous study, we demonstrated that a single immunization with H5 HA DNA vaccine, combined with electroporation, elicited both humoral and cellular immune responses, which provided mice with early protection against avian influenza H5N1 virus [21]. The experiments described in the preceding sections suggest that the HA1 DNA vaccine induced a weaker immune response than the HA and HAe vaccines. Therefore, we investigated whether the HA1 DNA vaccine might also induce a weaker immune response 1 week after immunization. In this experiment, 6- to 8-week-old female BALB/c mice (n = 10 per group) were immunized with 50 μg of HA, HAe, or HA1 with high-voltage electroporation (100 V). The mice were immunized once and challenged 7 days later. Sera were collected 7 days after immunization, and an antibody analysis showed that the mice in all immunized groups had a relatively lower antibody response (Fig. 4a–c). Similarly, the HA1 DNA vaccine also induced lower IgG, HAI, and neutralizing antibody titers than the HA and HAe DNA vaccines, although the differences were not significant (Fig. 4b, c). After challenge, the mice in the HA- and HAe-immunized groups all survived for 2 weeks (Fig. 4d), whereas only 80 % (4/5) of the mice in the HA1-immunized group survived. At 3 dpi, viral infection caused a weight loss of approximately 20 % in each group of mice. However, the weight losses differed at 5 dpi and were approximately 11 %, 23 %, and 27 % in the HA, HAe, and HA1 groups, respectively. The mice in the HA1 group also showed a more significant loss in bodyweight than the mice in the HA and HAe groups (p < 0.01). The weights of the HA- and HAe-treated mice recovered to the preinfection level after 2 weeks, but the weights of the HA1-immunized mice did not recover to the preinfection level (Fig. 4e). Consistent with weight loss, a high viral load was detected in the immunized mouse group and was similar to that in the control group (Fig. 4f). This result indicates that immunization with the HA1 DNA vaccine, with a challenge 1 week later, produced a weaker immune response and less protection than the HA and HAe DNA vaccines, as indicated by the relative antibody titers, survival rates, and bodyweight losses.

BALB/c mice were immunized once with 50 μg of pCAGGSP7/HA, pCAGGSP7/HAe, or pCAGGSP7/HA1 with high-voltage electroporation (100 V). Seven days after immunization, the sera were collected for the titration of IgG, HAI, and NAbs. a Titers of IgG in mouse serum after boosting. b HAI titers in vaccinated mice. c NAb titers in vaccinated mice. Seven days after immunization, the mice were challenged with a lethal dose of mouse-adapted NYMC X-179A (10 × LD50). Survival rates, bodyweight changes, and lung viral titers were determined. d Percentage survival of mice infected with lethal virus. e Bodyweight losses. f Lung viral titers on day 3 post-challenge
Discussion
DNA vaccines constitute a powerful alternative to conventional vaccines because they can induce both humoral and cellular immune responses. Many studies have shown that H1, H3, H5, H7, and H9 DNA vaccines can protect animals against viral infection [24, 40, 48–50]. In this study, we compared the immunogenicity of different forms of HA in mice. Our results show that two immunizations with HA, HAe, or HA1 DNA, combined with electroporation at a voltage of 100 V, induced strong immune responses against the HA protein of the H1N1pdm09 virus. Protection against lethal viral challenge was also provided by this DNA immunization regimen. The viral titers were extremely low or undetectable in the lungs of the immunized mice at 3 dpi (Fig. 2d–f), suggesting that the DNA vaccines we designed and tested effectively prevented H1N1pdm09 infection in mice. The HA1 DNA induced relatively lower IgG antibody, HAI, and neutralizing antibody titers than the HA or HAe DNA (Fig. 2a–c). The differences in the antibody responses observed in this study are consistent with the results reported previously by Tongawa et al. [43] and may be predominantly attributable to the fact that HA1 lacks the stalk domain of HA, which we deliberately omitted during the construction of the plasmid and has been shown to elicit a protective immune response [51]. Another explanation that cannot be excluded is the different cellular localization of HA, HAe, and HA1 (Fig. 1b), which may have caused different immune responses to be induced in the mice. Several studies support this inference [52, 53].
Electroporation following intramuscular injection markedly enhanced the efficacy of immunization. However, it has been shown that electroporation can damage the muscular tissue and cause an inflammatory reaction [54]. During electroporation, animals often feel discomfort, generated by myospasm. Higher voltages usually produce more-severe lesions [54]. As a further test of the immunogenicity of HA, HAe, and HA1, we assessed their ability to induce an immune response after electroporation at a voltage of 10 V. The antibody titers induced in the mice decreased as the voltage decreased from 100 V to 10 V, a phenomenon observed in our previous study [47]. Although the HA1 DNA induced a protective immune response against lethal viral infection, the IgG antibody, HAI, and neutralizing antibody titers elicited by it were lower than those elicited by the HA or HAe DNA. Consistent with these different antibody responses, mice immunized with HA1 showed greater weight loss and a higher viral load in lung washes after challenge. The HA1 subunit contains important neutralizing epitopes and is the region that predominantly induces the neutralizing activity against influenza virus infections. Some studies have suggested that it can be used for the development of a subunit influenza vaccine [55, 56]. However, in contrast to our results, Khurana et al. [31] reported that bacterially expressed HA1 protein induced higher titers of neutralizing antibodies than HAe or HA in rabbits and sheep. A possible explanation for the discrepancies in these studies could be that the bacterially expressed HA1 globular domain, but not HAe or HA, contains functional structures required for fetuin binding and red blood cell agglutination [31]. Therefore, the bacterially expressed recombinant proteins have folded structures that are altered during purification. With DNA vaccinations, because the encoded protein is synthesized in its native form inside the host cell [57], the antibody responses induced in mice by HA, HAe, or HA1 DNA vaccination in our study reflected the real immunogenicity of the antigen in mice.
The HA, HAe, and HA1 DNA vaccinations induced early immune responses, as demonstrated by the IgG, HAI, and neutralizing titers, although only low-level antibodies were detected at that time (Fig. 4). These results are consistent with our previous study, in which Zheng et al. [21] observed an early but low-level antibody response in mice immunized with H5 HA DNA. Although only low-level antibody responses were induced, >80 % of the mice immunized with HA, HAe, or HA1 survived a lethal viral challenge. Thus, in the absence or presence of low-level neutralizing antibodies, DNA immunization induced early protection against viral infection, which has also been observed in other studies. McLauchlan et al. [58] showed that a dose of DNA induced protection in fish challenged at either 1 or 4 weeks after vaccination. A similar result was observed in a horse model, in which the authors showed that a single dose of vaccine provided early protection against the development of viremia after challenge, even in the absence of measurable antibody titers in some horses [59]. In addition to low-level neutralizing antibodies, the cellular immune response may be involved in mediating this early protection. In our previous study [21], mice immunized once with HA DNA raised significant amounts of interferon-γ-producing cells in response to stimulation with the H5 HA protein. The present study also showed that the HA1 DNA vaccine induced lower IgG, HAI, and neutralization titers than the HA and HAe vaccines (Fig. 4b, c). Viral infection caused greater weight loss in mice immunized with HA1, and their bodyweights did not recover to their preinfection levels (Fig. 4e). These results confirm that HA1 induced less protective immunity than HA or HAe, as indicated by their antibody titers, survival rates, and bodyweight losses.
In summary, we have compared the capacities of HA, HAe, and HA1 to induce an antibody response and protection against viral infection. The HA1 DNA vaccine induced relatively weaker antibody responses and protective effects in mice than the HA and HAe DNA vaccines. These results extend our knowledge of the immunogenicity of different forms of HA protein in mice, which should be considered when developing vaccines to control influenza.
References
Ginsberg M, Hopkins J, Maroufi A, Dunne G, Sunega DR, Giessick J, McVay P, Lopez K, Kriner P, Lopez K, Munday S, Harriman K, Sun B, Chavez G, Hatch D, Schechter R, Vugia D, Louie J, Pascoe N, Penfield S, Zoretic J, Fonseca V, Blair P, Faix D, Tueller J, Gomez T, Averhoff F, Alavrado-Ramy F, Waterman S, Neatherlin J, Finelli L, Jain S, Brammer L, Bresee J, Bridges C, Doshi S, Donis R, Garten R, Katz J, Klimov S, Jernigan D, Lindstrom S, Shu B, Uyeki T, Xu X, Cox N (2009) Swine influenza A (H1N1) infection in two children—Southern California, March–April 2009. MMWR Morb Mortal Wkly Rep 58(15):400–402
World Healthy Organization (2009) Pandemic (H1N1)—update 68. http://www.who.int/csr/don/2009_10_02/en/index.html
Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, Lopez-Gatell H, Olivera H, Lopez I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov AI, Cox NJ (2009) Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325(5937):197–201
Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A (2009) Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459(7250):1122–1125
Dawood FS, Jain S, Finelli L, Shaw MW, Lindstrom S, Garten RJ, Gubareva LV, Xu X, Bridges CB, Uyeki TM (2009) Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N Engl J Med 360(25):2605–2615
Han K, Zhu X, He F, Liu L, Zhang L, Ma H, Tang X, Huang T, Zeng G, Zhu BP (2009) Lack of airborne transmission during outbreak of pandemic (H1N1) 2009 among tour group members, China. Emerg Infect Dis 15(10):1578–1581
Fraser C, Donnelly CA, Cauchemez S, Hanage WP, Van Kerkhove MD, Hollingsworth TD, Griffin J, Baggaley RF, Jenkins HE, Lyons EJ, Jombart T, Hinsley WR, Grassly NC, Balloux F, Ghani AC, Ferguson NM, Rambaut A, Pybus OG, Lopez-Gatell H, Alpuche-Aranda CM, Chapela IB, Zavala EP, Guevara DM, Checchi F, Garcia E, Hugonnet S, Roth C (2009) Pandemic potential of a strain of influenza A (H1N1): early findings. Science 324(5934):1557–1561
Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai-Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki-Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman-Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y (2009) In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature 460(7258):1021–1025
Maines TR, Jayaraman A, Belser JA, Wadford DA, Pappas C, Zeng H, Gustin KM, Pearce MB, Viswanathan K, Shriver ZH, Raman R, Cox NJ, Sasisekharan R, Katz JM, Tumpey TM (2009) Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science 325(5939):484–487
Munster VJ, de Wit E, van den Brand JM, Herfst S, Schrauwen EJ, Bestebroer TM, van de Vijver D, Boucher CA, Koopmans M, Rimmelzwaan GF, Kuiken T, Osterhaus AD, Fouchier RA (2009) Pathogenesis and transmission of swine-origin 2009 A(H1N1) influenza virus in ferrets. Science 325(5939):481–483
Hancock K, Veguilla V, Lu X, Zhong W, Butler EN, Sun H, Liu F, Dong L, DeVos JR, Gargiullo PM, Brammer TL, Cox NJ, Tumpey TM, Katz JM (2009) Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. N Engl J Med 361(20):1945–1952
Lim S, Kapell D, Zimmerman C, Nguyen T (2009) Effectiveness of 2008–09 trivalent influenza vaccine against 2009 pandemic influenza A (H1N1)—United States, May–June 2009. MMWR Morb Mortal Wkly Rep 58(44):1241–1245
Greenberg ME, Lai MH, Hartel GF, Wichems CH, Gittleson C, Bennet J, Dawson G, Hu W, Leggio C, Washington D, Basser RL (2009) Response to a monovalent 2009 influenza A (H1N1) vaccine. N Engl J Med 361(25):2405–2413
Clark TW, Pareek M, Hoschler K, Dillon H, Nicholson KG, Groth N, Stephenson I (2009) Trial of 2009 influenza A (H1N1) monovalent MF59-adjuvanted vaccine. N Engl J Med 361(25):2424–2435
Zhu FC, Wang H, Fang HH, Yang JG, Lin XJ, Liang XF, Zhang XF, Pan HX, Meng FY, Hu YM, Liu WD, Li CG, Li W, Zhang X, Hu JM, Peng WB, Yang BP, Xi P, Wang HQ, Zheng JS (2009) A novel influenza A (H1N1) vaccine in various age groups. N Engl J Med 361(25):2414–2423
Liang XF, Wang HQ, Wang JZ, Fang HH, Wu J, Zhu FC, Li RC, Xia SL, Zhao YL, Li FJ, Yan SH, Yin WD, An K, Feng DJ, Cui XL, Qi FC, Ju CJ, Zhang YH, Guo ZJ, Chen PY, Chen Z, Yan KM, Wang Y (2010) Safety and immunogenicity of 2009 pandemic influenza A H1N1 vaccines in China: a multicentre, double-blind, randomised, placebo-controlled trial. Lancet 375(9708):56–66
Kodihalli S, Goto H, Kobasa DL, Krauss S, Kawaoka Y, Webster RG (1999) DNA vaccine encoding hemagglutinin provides protective immunity against H5N1 influenza virus infection in mice. J Virol 73(3):2094–2098
Chen J, Fang F, Li X, Chang H, Chen Z (2005) Protection against influenza virus infection in BALB/c mice immunized with a single dose of neuraminidase-expressing DNAs by electroporation. Vaccine 23(34):4322–4328
Ogunremi O, Pasick J, Kobinger GP, Hannaman D, Berhane Y, Clavijo A, van Drunen Littel-van den Hurk S (2013) A single electroporation delivery of a DNA vaccine containing the hemagglutinin gene of Asian H5N1 avian influenza virus generated a protective antibody response in chickens against a North American virus strain. Clin Vaccine Immunol 20(4):491–500
Chen Q, Kuang H, Wang H, Fang F, Yang Z, Zhang Z, Zhang X, Chen Z (2009) Comparing the ability of a series of viral protein-expressing plasmid DNAs to protect against H5N1 influenza virus. Virus Genes 38(1):30–38
Zheng L, Wang F, Yang Z, Chen J, Chang H, Chen Z (2009) A single immunization with HA DNA vaccine by electroporation induces early protection against H5N1 avian influenza virus challenge in mice. BMC Infect Dis 9:17
Hatta M, Gao P, Halfmann P, Kawaoka Y (2001) Molecular basis for high virulence of Hong Kong H5N1 influenza A viruses. Science 293(5536):1840–1842
Van Hoeven N, Pappas C, Belser JA, Maines TR, Zeng H, Garcia-Sastre A, Sasisekharan R, Katz JM, Tumpey TM (2009) Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. Proc Natl Acad Sci USA 106(9):3366–3371
Lee CW, Senne DA, Suarez DL (2006) Development and application of reference antisera against 15 hemagglutinin subtypes of influenza virus by DNA vaccination of chickens. Clin Vaccine Immunol 13(3):395–402
Jones S, Evans K, McElwaine-Johnn H, Sharpe M, Oxford J, Lambkin-Williams R, Mant T, Nolan A, Zambon M, Ellis J, Beadle J, Loudon PT (2009) DNA vaccination protects against an influenza challenge in a double-blind randomised placebo-controlled phase 1b clinical trial. Vaccine 27(18):2506–2512
Shen S, Mahadevappa G, Oh HL, Wee BY, Choi YW, Hwang LA, Lim SG, Hong W, Lal SK, Tan YJ (2008) Comparing the antibody responses against recombinant hemagglutinin proteins of avian influenza A (H5N1) virus expressed in insect cells and bacteria. J Med Virol 80(11):1972–1983
Prabakaran M, Velumani S, He F, Karuppannan AK, Geng GY, Yin LK, Kwang J (2008) Protective immunity against influenza H5N1 virus challenge in mice by intranasal co-administration of baculovirus surface-displayed HA and recombinant CTB as an adjuvant. Virology 380(2):412–420
Toro H, Tang DC (2009) Protection of chickens against avian influenza with nonreplicating adenovirus-vectored vaccine. Poult Sci 88(4):867–871
Kreijtz JH, Suezer Y, de Mutsert G, van den Brand JM, van Amerongen G, Schnierle BS, Kuiken T, Fouchier RA, Lower J, Osterhaus AD, Sutter G, Rimmelzwaan GF (2009) Preclinical evaluation of a modified vaccinia virus Ankara (MVA)-based vaccine against influenza A/H5N1 viruses. Vaccine 27(45):6296–6299
Barefoot BE, Athearn K, Sample CJ, Ramsburg EA (2009) Intramuscular immunization with a vesicular stomatitis virus recombinant expressing the influenza hemagglutinin provides post-exposure protection against lethal influenza challenge. Vaccine 28(1):79–89
Khurana S, Verma S, Verma N, Crevar CJ, Carter DM, Manischewitz J, King LR, Ross TM, Golding H (2010) Properly folded bacterially expressed H1N1 hemagglutinin globular head and ectodomain vaccines protect ferrets against H1N1 pandemic influenza virus. PLoS One 5(7):e11548
Lardinois A, Steensels M, Lambrecht B, Desloges N, Rahaus M, Rebeski D, van den Berg T (2012) Potency of a recombinant NDV-H5 vaccine against various HPAI H5N1 virus challenges in SPF chickens. Avian Dis 56(4 Suppl):928–936
Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P (2007) A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog 3(10):1414–1421
Shim BS, Choi JA, Song HH, Park SM, Cheon IS, Jang JE, Woo SJ, Cho CH, Song MS, Kim H, Song KJ, Lee JM, Kim SW, Song DS, Choi YK, Kim JO, Nguyen HH, Kim DW, Bahk YY, Yun CH, Song MK (2013) Sublingual administration of bacteria-expressed influenza virus hemagglutinin 1 (HA1) induces protection against infection with 2009 pandemic H1N1 influenza virus. J Microbiol 51(1):130–135
Steel J, Lowen AC, Mubareka S, Palese P (2009) Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627 K or 627E/701 N. PLoS Pathog 5(1):e1000252
Herfst S, Chutinimitkul S, Ye J, de Wit E, Munster VJ, Schrauwen EJA, Bestebroer TM, Jonges M, Meijer A, Koopmans M, Rimmelzwaan GF, Osterhaus ADME, Perez DR, Fouchier RAM (2010) Introduction of virulence markers in PB2 of pandemic swine-origin influenza virus does not result in enhanced virulence or transmission. J Virol 84(8):3752–3758
Reed LJ, Muench H (1938) A simple method of estimating fifty per cent endpoints. Am J Hyg 27:493–497
Chen Z, Sahashi Y, Matsuo K, Asanuma H, Takahashi H, Iwasaki T, Suzuki Y, Aizawa C, Kurata T, Tamura S (1998) Comparison of the ability of viral protein-expressing plasmid DNAs to protect against influenza. Vaccine 16(16):1544–1549
Aihara H, Miyazaki J (1998) Gene transfer into muscle by electroporation in vivo. Nat Biotechnol 16(9):867–870
Qiu M, Fang F, Chen Y, Wang H, Chen Q, Chang H, Wang F, Zhang R, Chen Z (2006) Protection against avian influenza H9N2 virus challenge by immunization with hemagglutinin- or neuraminidase-expressing DNA in BALB/c mice. Biochem Biophys Res Commun 343(4):1124–1131
Mozdzanowska K, Furchner M, Washko G, Mozdzanowski J, Gerhard W (1997) A pulmonary influenza virus infection in SCID mice can be cured by treatment with hemagglutinin-specific antibodies that display very low virus-neutralizing activity in vitro. J Virol 71(6):4347–4355
Li X, Fang F, Song Y, Yan H, Chang H, Sun S, Chen Z (2006) Essential sequence of influenza neuraminidase DNA to provide protection against lethal viral infection. DNA Cell Biol 25(4):197–205
Tonegawa K, Nobusawa E, Nakajima K, Kato T, Kutsuna T, Kuroda K, Shibata T, Harada Y, Nakamura A, Itoh M (2003) Analysis of epitope recognition of antibodies induced by DNA immunization against hemagglutinin protein of influenza A virus. Vaccine 21(23):3118–3125
Torres CA, Yang K, Mustafa F, Robinson HL (1999) DNA immunization: effect of secretion of DNA-expressed hemagglutinins on antibody responses. Vaccine 18(9–10):805–814
Ilyushina NA, Khalenkov AM, Seiler JP, Forrest HL, Bovin NV, Marjuki H, Barman S, Webster RG, Webby RJ (2010) Adaptation of pandemic H1N1 influenza viruses in mice. J Virol 84(17):8607–8616
Ye J, Sorrell EM, Cai Y, Shao H, Xu K, Pena L, Hickman D, Song H, Angel M, Medina RA, Manicassamy B, Garcia-Sastre A, Perez DR (2010) Variations in the hemagglutinin of the 2009 H1N1 pandemic virus: potential for strains with altered virulence phenotype? PLoS Pathog 6(10):e1001145
Zhou Y, Fang F, Chen J, Wang H, Chang H, Yang Z, Chen Z (2008) Electroporation at low voltages enables DNA vaccine to provide protection against a lethal H5N1 avian influenza virus challenge in mice. Intervirology 51(4):241–246
Laddy DJ, Yan J, Corbitt N, Kobasa D, Kobinger GP, Weiner DB (2007) Immunogenicity of novel consensus-based DNA vaccines against avian influenza. Vaccine 25(16):2984–2989
Ljungberg K, Kolmskog C, Wahren B, van Amerongen G, Baars M, Osterhaus A, Linde A, Rimmelzwaan G (2002) DNA vaccination of ferrets with chimeric influenza A virus hemagglutinin (H3) genes. Vaccine 20(16):2045–2052
Cherbonnel M, Rousset J, Jestin V (2003) Strategies to improve protection against low-pathogenicity H7 avian influenza virus infection using DNA vaccines. Avian Dis 47(3 Suppl):1181–1186
Steel J, Lowen AC, Wang TT, Yondola M, Gao Q, Haye K, Garcia-Sastre A, Palese P (2010) Influenza virus vaccine based on the conserved hemagglutinin stalk domain. MBio 1(1)
Lewis PJ, van Drunen Littel-van den H, Babiuk LA (1999) Altering the cellular location of an antigen expressed by a DNA-based vaccine modulates the immune response. J Virol 73(12):10214–10223
Kaur R, Sachdeva G, Vrati S (2002) Plasmid DNA immunization against Japanese encephalitis virus: immunogenicity of membrane-anchored and secretory envelope protein. J Infect Dis 185(1):1–12
Babiuk S, Baca-Estrada ME, Foldvari M, Storms M, Rabussay D, Widera G, Babiuk LA (2002) Electroporation improves the efficacy of DNA vaccines in large animals. Vaccine 20(27–28):3399–3408
Du L, Leung VH, Zhang X, Zhou J, Chen M, He W, Zhang HY, Chan CC, Poon VK, Zhao G, Sun S, Cai L, Zhou Y, Zheng BJ, Jiang S (2011) A recombinant vaccine of H5N1 HA1 fused with foldon and human IgG Fc induced complete cross-clade protection against divergent H5N1 viruses. PLoS One 6(1):e16555
Khurana S, Verma S, Verma N, Crevar CJ, Carter DM, Manischewitz J, King LR, Ross TM, Golding H (2011) Bacterial HA1 vaccine against pandemic H5N1 influenza virus: evidence of oligomerization, hemagglutination, and cross-protective immunity in ferrets. J Virol 85(3):1246–1256
Gurunathan S, Klinman DM, Seder RA (2000) DNA vaccines: immunology, application, and optimization. Annu Rev Immunol 18:927–974
McLauchlan PE, Collet B, Ingerslev E, Secombes CJ, Lorenzen N, Ellis AE (2003) DNA vaccination against viral haemorrhagic septicaemia (VHS) in rainbow trout: size, dose, route of injection and duration of protection-early protection correlates with Mx expression. Fish Shellfish Immunol 15(1):39–50
Siger L, Bowen RA, Karaca K, Murray MJ, Gordy PW, Loosmore SM, Audonnet JC, Nordgren RM, Minke JM (2004) Assessment of the efficacy of a single dose of a recombinant vaccine against West Nile virus in response to natural challenge with West Nile virus-infected mosquitoes in horses. Am J Vet Res 65(11):1459–1462
Acknowledgments
This study was supported by the following research funds: National 973 Project (2010CB530301); National Natural Science Foundation of China (310000088); Foundation for Study Encouragement to Young Scientists, Chinese Academy of Sciences (KSCX2-EW-J-19) and The Ministry of Science and Technology Special Project (2013FY113500).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Chen, J., Liu, Q., Chen, Q. et al. Comparative analysis of antibody induction and protection against influenza virus infection by DNA immunization with HA, HAe, and HA1 in mice. Arch Virol 159, 689–700 (2014). https://doi.org/10.1007/s00705-013-1878-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-013-1878-1