
Abstract
The claim that it is possible to rationally design a structure-based HIV-1 vaccine is based on misconceptions regarding the nature of protein epitopes and of immunological specificity. Attempts to use reverse vaccinology to generate an HIV-1 vaccine on the basis of the structure of viral epitopes bound to monoclonal neutralizing antibodies have failed so far because it was not possible to extrapolate from an observed antigenic structure to the immunogenic structure required in a vaccine. Vaccine immunogenicity depends on numerous extrinsic factors such as the host immunoglobulin gene repertoire, the presence of various cellular and regulatory mechanisms in the immunized host and the process of antibody affinity maturation. All these factors played a role in the appearance of the neutralizing antibody used to select the epitope to be investigated as potential vaccine immunogen, but they cannot be expected to be present in identical form in the host to be vaccinated. It is possible to rationally design and optimize an epitope to fit one particular antibody molecule or to improve the paratope binding efficacy of a monoclonal antibody intended for passive immunotherapy. What is not possible is to rationally design an HIV-1 vaccine immunogen that will elicit a protective polyclonal antibody response of predetermined efficacy. An effective vaccine immunogen can only be discovered by investigating experimentally the immunogenicity of a candidate molecule and demonstrating its ability to induce a protective immune response. It cannot be discovered by determining which epitopes of an engineered antigen molecule are recognized by a neutralizing monoclonal antibody. This means that empirical immunogenicity trials rather than structural analyses of antigens offer the best hope of discovering an HIV-1 vaccine.
Similar content being viewed by others

Introduction
Human immunodeficiency virus type 1 (HIV-1) is the virus we know most about [1] and its antigenic structure has been analyzed in considerable detail [2–6]. Many studies have been devoted to the HIV-1 envelope glycoprotein (Env) present as spikes embedded in the viral membrane. Each functional spike consists of trimers of three gp120 surface glycoproteins non-covalently attached to three gp41 transmembrane glycoproteins. In addition to functional trimers, the viral membrane may also display gp120-gp41 monomers, single gp41 molecules and other non-functional fragments [7–9]. The antigenicity of the spikes has been studied extensively and numerous Env epitopes have been shown to induce a wide range of neutralizing antibodies in HIV-1 infected individuals [4, 10–17]. As a result, the antigenic sites of the Env protein have been considered promising candidates for developing a preventive HIV vaccine able to provide prophylactic immunity [18–22].
Many studies have been undertaken to determine the structure of epitopes bound to neutralizing monoclonal antibodies (Mabs) in the hope that such epitopes could be turned into effective vaccine immunogens [23, 24]. The strategy of trying to develop a vaccine by studying the interaction of human Mabs with HIV-1 Env epitopes has been called reverse vaccinology because it attempts to generate a vaccine from the known structure of neutralizing antibodies rather than by following the reverse, more common approach of generating protective antibodies by immunization [23, 24]. When crystallographers study the structure of an Env epitope recognized by a neutralizing Mab, they often claim that their data will help the rational design of vaccine immunogens able to elicit antibodies endowed with the same neutralizing capacity as the Mab used to define the epitope [25–27].
In vaccine development, the term design usually means selecting a vaccine candidate and finding appropriate vaccine formulations, schedules, adjuvants and routes of administration. The strategy of rational, structure-based design discussed here is more circumscribed since it refers to the development and optimization of a vaccine immunogen within the restricted context of a single Mab-antigen pair. This approach assumes that if a viral antigen can be modified so that it binds better to a neutralizing Mab, it will have acquired the immunogenic capacity of eliciting neutralizing polyclonal antibodies [28].
It will be argued here that the claims of reverse vaccinology are based (1) on misconceptions regarding the nature of protein epitopes and of immunological specificity, (2) on the assumption that a discontinuous epitope defined by X-ray crystallography can act as an effective vaccine immunogen on its own without being embedded in a multi-epitopic protein that always induces many different types of antibodies, (3) on the belief that effective vaccine immunogens can be predicted from the antigenic structure of viral antigens bound to neutralizing Mabs.
Research carried out over the last two decades has demonstrated that reverse vaccinology has not been able to guide the design of an HIV vaccine nor of any other viral vaccine [29]. The present review expands on an earlier analysis of the limitations of reverse vaccinology [29] and shows that antibody polyspecificity explains why elucidating the structure of viral epitopes recognized by a neutralizing Mab is of little use for discovering immunogens capable of inducing a protective immune response. It may be advisable, therefore, to shift the current emphasis in HIV vaccine research from a rational design approach based on the structural analysis of Env epitopes to a more empirical approach that investigates possible therapeutic benefits that may result from the administration of a variety of HIV-1 vaccine immunogens [30].
The nature of protein epitopes
The regions of antigen molecules recognized by antibody molecules are called epitopes while the regions of antibodies that bind to epitopes are called paratopes. Both regions are identified by solving the 3D structure of antigen-antibody complexes and determining which amino acid residues in the two partners make contact with each other.
Discontinuous epitopes
The vast majority of protein epitopes have been found to be discontinuous [31–33] and to consist of two to five short stretches of residues that are distant in the protein sequence but are brought together by the folding of the peptide chain. Most HIV-1 epitopes that have been characterized are discontinuous epitopes [34–37]. The atomic groups forming a discontinuous epitope are not held together by internal chemical bonds and the epitope is a 3D entity that emerges only because the chain acts as a scaffold. If the scaffold is perturbed, for instance by a change in chain conformation or by denaturation of the protein, the epitope ceases to exist [38, 39]. Discontinuous epitopes cannot be extracted and isolated in active form from the protein antigen and they possess binding activity only within the context of the 3D protein structure in which they are embedded (Fig. 1). In spite of claims to the contrary, it is not possible to predict the exact structure of a discontinuous epitope from nucleotide or amino acid sequences [40–43].

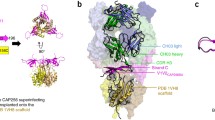
Discontinuous epitope of the outer surface protein A of the spirochete Borrelia burdorferi elucidated by X-ray crystallography from a complex with Mab 184.1. (A) Outline of the epitope in yellow, (B) Position in space of the residues comprising the epitope. This set of residues cannot be isolated as such from the protein to demonstrate that it possesses binding activity in its own. (C) Parts of the discontinuous epitope and other peptide segments of the protein that may be able to bind Mab 184.1, in which case they would be called continuous epitopes (courtesy of Pernille Haste-Andersen, Danish Technical University)
Continuous epitopes
A second type of protein epitope known as a continuous epitope is defined as any short peptide fragment of the protein, usually five to eight residues long, that is able to cross-react with antibodies raised against the protein. When linear peptides are given the status of continuous epitopes of a protein, the impression is created that these epitopes actually exist as discrete binding and immunogenic sites in the native protein. In most cases, however, only some of the residues of a continuous epitope are located at the surface of the native protein where they are usually part of a more complex discontinuous epitope (Fig. 1). Although epitope databases list thousands of continuous epitopes presumed to exist in individual proteins [44, 45], these linear peptides are actually poor mimics of the larger antigenic structures present in native proteins and they mostly cross-react only weakly with antiprotein antibodies [46]. When the structure of continuous epitopes bound to anti-peptide antibodies is compared to the structure of the corresponding regions in the cognate native protein, the two structures are usually found to differ considerably, with the result that most antibodies raised against the peptides are unable to bind to the parent protein because bumps and clashes prevent antibody binding [47].
Continuous epitopes always have ill-defined, fuzzy boundaries because they are only defined by their binding activity rather than by showing that all the residues in the peptide interact with antiprotein antibodies [46]. Attempts are sometimes made to assess the boundaries of continuous epitopes by measuring the binding activity of peptides of decreasing size and giving the status of epitope to the smallest peptide that retains detectable activity. However, the results of such analyses depend on the immunoassay used because the peptide conformation usually differs in different types of assay [48]. In some assays, tripeptides can be shown to possess significant binding activity [49]. The contribution of individual residues to the epitope activity can also be evaluated by measuring the binding capacity of sets of peptide analogs presenting single residue replacements. When this is done, certain residues are found to be essential for binding while others, called replaceable residues, can be replaced by any of the other 19 amino acids without impairing antigenic reactivity [50, 51]. When a residue substitution is found to affect the epitope activity, it could mean that the residue is in contact with the paratope or that the substitution induced a conformational change which affects the reactivity of the peptide.
It has been suggested that most continuous epitopes are not genuine epitopes of native proteins but correspond to unfolded regions in denatured proteins that are recognized by antibodies directed to denatured molecules [38]. It can, indeed, never be excluded that the antiprotein antiserum used for detecting continuous epitopes contains antibodies specific for denatured protein molecules because some of the protein molecules used for immunization became denatured before or after being injected in the animal [52]. In the reciprocal situation where antibodies raised to peptides are allowed to react with the cognate protein, it also cannot be excluded that the antibodies recognize denatured protein molecules present in the immunoassay format that was used [53]. It is now accepted, for instance, that earlier claims that immunization with peptides always elicits high levels of antibodies that cross-react with the native cognate protein [54] arose because it was not realized at the time that the protein used in solid-phase immunoassays became denatured by adsorption to plastic [46, 55]. The classification of epitopes as either continuous or discontinuous may give the impression that the fundamental units of antigenic recognition are amino acid residues, although epitope-paratope interactions involve only a few atoms in each residue which may be side chain or main chain atoms. It should also be appreciated that various crystallographic groups often use different criteria for identifying which contact residues are involved in an interface [50]. For instance, contact residues in the epitope have been defined as those that have any atom less than 0.5 nm away from any atom in the antibody [56]. Other distance thresholds have been used for identifying different types of contact, for instance 0.35 nm for hydrogen bonds and salt bridges, 0.4 nm for van der Waals interactions and 0.45 nm for π cation interactions [47]. The use of different thresholds may lead to different residues being attributed to the binding interface.
Neotopes and cryptotopes
The quaternary structure of the protein subunits assembled in virus particles gives rise to additional types of epitopes known as neotopes and cryptotopes. The term neotope was introduced in 1966 [57] to refer to epitopes that arise either from the juxtaposition of residues in neighbouring proteins or subunits that are recognized by antibodies as a single epitope, or from conformational changes induced in the proteins by intersubunit interactions [58]. Neotopes have been found in the capsid and membrane proteins of most viruses [59, 60] and are also present in HIV-1 [61–63]. Since the quaternary structure of virions is dynamic rather than static [64, 65], neotopes tend to be transient epitopes that can assume different conformations [4, 21].
When the glycoproteins in HIV-1 spikes associate to form trimers, a part of their surface is no longer accessible to antibodies. These masked regions harbour epitopes known as cryptotopes [66] that are active only when the subunits are dissociated [61].
There is evidence that differences in the quaternary structure of gp120 between primary HIV-1 isolates, i.e. viruses that have been minimally passaged in cell lines, and T-cell line adapted (TCLA) isolates are responsible for their differential sensitivity to neutralization. It seems that a more open gp 120 conformation in TCLA viruses allows neutralizing antibodies to access more easily the binding sites for CD4 and for the coreceptors CCR5 and CXCR4 as well as the V3 loop [13, 67, 68].
Mimotopes
Another type of linear peptide epitope was recognized in 1986 and given the label mimotope [69]. Mimotopes are detected when peptides from combinatorial peptide libraries obtained by chemical synthesis or phage display are tested for their ability to bind antiprotein Mabs that recognize either continuous or discontinuous epitopes [70]. When the Mabs are directed to discontinuous epitopes, they may bind to peptides showing little or no sequence similarity with the antigen used to elicit the antiprotein antibodies. Antigenic cross-reactions occurring in the absence of sequence similarities may arise from the overriding attraction between hydrophilic and hydrophobic groups that occurs when a pattern of hydropathic complementarity is conserved in dissimilar peptide sequences [71, 72]. Such a phenomenon is responsible for the binding of complementary peptides specified by the sense and antisense strands of DNA molecules because their respective codons always code for amino acids of opposite hydropathicity [73, 74].
When Mabs used to detect mimotopes recognize a continuous epitope in a protein, there is sometimes extensive sequence identity between the mimotope and the continuous epitope. In the case of two neutralizing Mabs that recognized the same 7-residue sequence present in the surface glycoprotein of a murine coronavirus and in several mimotopes, it was found that the two Mabs presented little similarity in their heavy chain complementarity determining regions (CDRs), indicating that two paratopes that recognize similar sequences can be very different [75]. It is also often found that Mabs harbouring unrelated paratopes are able to recognize very similar discontinuous epitopes [76–78], suggesting that it may be difficult to control the type of antibody that will be obtained when very similar epitopes are used as immunogens.
A limited number of mimotopes may also be able to induce antibodies that cross-react with the protein [79–81] and show a modest capacity to neutralize viral infectivity [82, 83]. The antigenic and immunogenic activities of mimotopes often depend on their conformation. For instance, when mimotopes are displayed on a phage, they may better mimic the conformation of a viral epitope than when they are used as free peptides or conjugated to a carrier protein [70, 84, 85]. Mimotopes displayed on a phage protein may also be better elicitors of antibodies that cross-react with the virus and neutralize its infectivity [75]. Although several mimotopes of HIV-1 epitopes have been isolated from combinatorial peptide libraries [84–88] none of them were able, when used as immunogens, to elicit adequate levels of neutralizing and protective antibodies.
What do mimotopes mimic?
Many peptides that are given the label mimotope because they cross-react with an antiprotein Mab do not necessarily bind only to residues of the known antiprotein paratope since they may interact with neighbouring residues that are part of a different paratope recognized by the mimotope. As a result, many so-called mimotopes do not really mimic the epitope recognized by the Mab but simply react with polyspecific immunoglobulin (Ig) molecules that always possess numerous potential binding sites for different epitopes (see “The specificity of antibodies is limited rather than absolute”).
The most commonly used procedure for assessing what a mimotope recognizes is to test whether it is able to inhibit the binding of the Mab to its homologous antigen. If the mimotope is found to compete with the antigen for binding to the Mab, this is usually taken as sufficient evidence that it mimics the epitope recognized by the Mab [70, 84]. This criterion for establishing mimicry is actually questionable since the binding inhibition could simply be due to the fact that steric hindrance prevents the mimotope from binding to a neighbouring paratope in the Ig binding pocket of the Mab. If an antibody recognizes two unrelated epitopes through separate, non-overlapping paratopes, it would of course make little sense to say that the two epitopes are antigenic mimics of each other. If the mimotope presents some sequence similarity with portions of the epitope, this is taken as evidence that the mimotope binds to the putative paratope of the Mab. However, it is equally plausible that the mimotope binds to its “own” specific site consisting of a neighbouring set of residues. Furthermore, if the mimotope is able to elicit antibodies that recognize the epitope believed to be mimicked, this is taken as confirming evidence that the mimotope exactly mimicks the epitope.
The antigenic mimicry displayed by a mimotope, however, does not imply that the peptide is a precise structural mimic making identical contacts with the paratope to those made by the epitope that elicited the antibody, since a modest similarity between two antigens may be sufficient to give rise to a cross-reaction involving overlapping but different paratopes. The concept of immunogenic mimicry is even more difficult to define in structural terms than antigenic mimicry, since it involves undefined functional activities in the immunized host as well as unknown characteristics of the immunogen that elicit the appearance of cross-reactive antibodies [70].
The specificity of antibodies is limited rather than absolute
The concept of specificity in immunology has been difficult to define [89–92]. The term specificity is derived from the word species and describes what is characteristic of a species [93]. For centuries, biological species were considered to be separated by clear-cut discontinuities and in the 19th century bacteriologists believed that antisera raised against different species of bacteria were completely specific, allowing different species to be identified by serology with absolute certainty [94]. This view was later questioned by Landsteiner [95] who showed that serological cross-reactions between different cell types were caused by antibodies that reacted to different degrees with a wide range of cells. He demonstrated that there was no one-to-one relationship between an antigen and its antibody and that an antigen could elicit a wide spectrum of antibodies capable of cross-reacting with many antigens. This led to antigen-antibody interactions being regarded as much less specific than previously thought and is nowadays explained by the widespread occurrence of cross-reactions between different epitopes and paratopes. The work of Landsteiner led Talmadge [96] to suggest that immunological specificity was not the result of a unique antibody molecule recognizing each distinguishable antigen but arose from the combinatorial effect of several different antibodies recognizing separate epitopes on the same antigen. According to that interpretation, a specific antigen recognition or elimination process is always initiated by several binding events involving different antibodies.
Specificity has been defined as “the exact complementary relationship between an agent and something acted on” [97], a definition which applies to the stereochemical complementarity observed between antigen and antibody, enzyme and substrate or receptor and ligand. In the case of antibodies, the recognition of a complementary antigen is mediated by the binding site of Ig molecules consisting of 50-70 hypervariable residues distributed over the six CDRs in the variable domains of the heavy of light Ig chains. Each immunoglobulin binding site can accommodate many overlapping or non-overlapping binding subsites of 10-20 residues, corresponding to paratopes able to bind different antigens. An Ig molecule may, for instance, possess two independent binding sites that allow it to bind simultaneously to phosphorylcholine and to α-D-galactopyranoside [98]. A more common situation is that different paratope subsites partly overlap, in which case binding to one epitope would prevent a second related or unrelated epitope from being accommodated at a nearby location in the Ig binding site. All antibody molecules are therefore polyspecific since they always harbor numerous subsites corresponding to paratopes specific for different epitopes. The flexibility of CDR loops also contributes to the ability of Igs to adapt to a variety of epitopes [99–101]. It is important to distinguish between an actual paratope and the virtual functional site of an Ig molecule consisting of all the residues of the six CDRs which, however, do not form a binding site for any particular epitope.
Degeneracy of the immune system and fuzzy recognition sites
Many terms such as cross-reactivity, molecular mimicry, plasticity, promiscuity, degeneracy and polyspecificity have been used to describe the finding that a T cell receptor (TCR) and B cell receptor (BCR) can recognize many different ligands [90, 102, 103]. The term polyspecificity is particularly fitting because it captures two essential features of immune receptors, i.e. their ability to recognize multiple peptide/MHC (major histocompatibility complex) ligands or B cell epitopes as well as their capacity to distinguish between closely related ligands that present only very small structural differences [104].
The term cross-reactivity does not imply that a particular mechanism is responsible for recognition but it suggests that there is a primary ligand for each TCR and BCR and that the binding of all other ligands are cases of cross-reaction. In reality, there is no single intrinsic or “real” epitope for any immune receptor but only a diverse group of potential ligands [104]. Cross-reactivity is also said to occur when an immune response and the resulting secreted antibodies directed at one antigen are able to interact with other antigens that did not stimulate the original reaction [103]. Cross-reactivity then refers to a lack of specificity.
Degeneracy is the ability of elements that are structurally different to perform the same activity or function. Degeneracy may yield the same or different functions depending on the context in which it is expressed [105]. The genetic code, for instance, is degenerate because there are many more triplet codons than encoded amino acid residues. As a result, an enormous number of structurally distinct mRNA species could be translated to generate the amino acid sequence of any particular protein. Degeneracy is further increased when one includes variations in polynucleotide sequences that result in mutated but functionally equivalent gene products [105]. In immunology, degeneracy refers not only to the ability of structurally different antibodies to perform the same recognition function but also to the ability of a single antibody to perform multiple functions like binding to different antigens. Degeneracy is a property of a system as a whole and not of its single components [103]. The degeneracy of T cell recognition refers to the fact that peptide-binding motifs are polyspecific for MHC class molecules and that a single TCR can be activated by a large number of different peptides possessing limited sequence similarity. The degeneracy of the immunoglobulins made by an animal ensures that the animal has the ability to make antibodies which recognize essentially any molecular motif which its immune system may encounter. If each immunological receptor could only recognize a single complementary ligand, the immune system would need one receptor for every possible epitope which would require orders of magnitude more receptors than there are cells in the immune system. It is only because of polyspecificity that a complete immune repertoire can be achieved by a much more restricted number of receptors [104]. Degeneracy is also a prerequisite to allow organisms to adapt over evolutionary time since natural selection can only operate among a population of dissimilar organisms [105].
The description of protein binding sites as fuzzy recognition sites was introduced by Moodie et al [106] to account for the finding that protein-ligand complementarity can be achieved by not just a single arrangement of amino acids but by a large number of alternative arrangements. Epitopes have also been called fuzzy recognition sites because there is no clear-cut minimum difference in binding affinity or in atomic positions at the antigen-antibody interface that can serve as an absolute yardstick for deciding that two epitopes are the same or not [93, 107]. The use of fuzziness as a descriptor of reality is useful for handling concepts and entities that do not possess clear-cut boundaries [108]. Whereas classical, Aristotelian logic analyses concepts and classes in terms of mutually exclusive dichotomies of the type: a glass is full or is not full and a statement is true or not true, fuzzy logic is a method to handle a continuum with innumerable steps [108–110]. Linguistic descriptors are often assumed to refer to entities that possess sharp boundaries because they satisfy the propensity of the human mind to want to make clear-cut distinctions. The continuous nature of the spectrum of electromagnetic waves does not prevent us from distinguishing different colors and the continuity of geological rock formations does not stop us from giving different names to individual mountain peaks. In a similar way, the continuous nature of biological variation and of protein evolution does not prevent us from distinguishing and giving separate names to individual biological species, antigenic binding sites and antibodies [93].
Even the classical tenet of immunological theory which posits the need of the immune system to discriminate between self and nonself [90], has been interpreted in terms of fuzzy recognition sites [111] and complex systems [112]. It used to be believed that T cells in the thymus were either clearly responsive or unresponsive to self and foreign antigens but there is now evidence that many self-reactive T cells which escape from clonal selection as a result of low affinity interactions with self-peptides, eventually become part of the general T cell repertoire [113]. The occurrence of antibodies able to react with foreign antigens results from selective forces operating during immunization which depend on reactant concentration and on the probability of receptor binding and lymphocyte stimulation occurring above a certain triggering threshold [114, 115].
Epitopes and paratopes are relational entities
Once an Ig molecule has been found to bind to an antigen, it becomes possible to solve the structure of the antigen-antibody complex and to identify the paratope and epitope regions. Epitopes and paratopes are actually relational entities defined by their mutual complementarity and they depend on each other to acquire a recognizable identity. An epitope is thus not an intrinsic structural feature of a protein that could be identified in the absence of a particular interaction with a paratope [39, 46]. This relational dependence means that as soon as the epitope is slightly altered and binding to the antibody is affected, the paratope also is no longer the same. As a result, analyzing the antigenicity of a protein amounts to analyzing the size of the immunological repertoire of the host immunized with that protein. The number of epitopes identifiable in a protein is therefore equal to the number of different Mabs that can be raised against it. Using this criterion, the insulin molecule was shown to possess 115 epitopes [116] and the BLysS molecule more than a thousand [117]. This analysis differs from the viewpoint advocated by Cohn [90] which defines a “paratopic clan” as a family of paratopes, distinguishable one from the other, that are all functionally capable to recognize a given single epitope. Cohn further defines a “mimotopic array” as a set of epitopes, distinguishable one from the other, that are all able to interact functionally with a given single paratope.
Epitopes and antigenic sites
It is now accepted that the entire accessible surface of a protein harbours a large number of overlapping epitopes that can be detected only if Mabs that bind all these regions are available [118, 119]. This means that the same residues at the surface of a protein can always be part of different overlapping epitopes. Even if the majority of residues recognized by two different Mabs are the same, the chemical bonding pattern between epitope and paratope may be very different and involve totally unrelated CDR sequences [78]. When only a small number of Mabs are available for studying the antigenicity of a protein, investigators tend to give undue importance to spurious boundaries assumed to demarcate discrete antigenic regions and they disregard the fact that the protein surface is an antigenic continuum. This leads to an analytical bias that emphasizes immune responses elicited by single epitopes rather than fostering an examination of the beneficial neutralizing synergy that often result from the combined interactions of several independent epitopes and paratopes [120–122]. Restricting one’s attention to single epitopes also negates the insight of Talmadge [96] that specific interactions result from the combined effect of several independent recognition events.
Although it is impossible to draw distinct boundaries between overlapping epitopes, it is customary to refer to the existence of separate antigenic sites in proteins, each one comprising many overlapping or non-overlapping epitopes. Each antigenic site is identified on the protein surface on the basis of a recognizable spatial location as well as by certain structural and functional properties. Several antigenic sites of HIV-1 Env spikes have been extensively studied, for instance the CD4 binding site (CD4bs) which is the region that allows the virus to attach to its primary CD4 receptor on the surface of target cells [16, 119, 123], the V3 antigenic site [4, 10, 17] and the membrane proximate external region (MPER) of gp41 [5, 11, 18].
Antibody heterospecificity
Antibody heterospecificity is the ability of an antibody to bind more strongly to a different antigen than the one used for raising the antibody and is frequently observed when an antibody is tested against a series of analogs or mutants of the immunogen [125–128]. Heterospecificity is due to the fact that the clonal selection of B cells, which subsequently leads to antibody secretion in plasma cells, can be triggered by an immunogen endowed with only moderate affinity for a B cell receptor. Antibodies of low or moderate affinity will then be obtained which may react better with heterologous epitopes endowed with a superior degree of complementarity with the paratope [39]. In some cases, antibodies may have such a low affinity for the immunogen that they will not react with it at all and will only bind to a mutated antigen presenting a substitution that facilitates heterospecific binding. The technique of intragel cross-absorption of antisera is particularly suitable for detecting this phenomenon [129]. It is well-known that following immunization with an antigen, antisera are obtained that contain levels of total immunoglobulins that far exceed the levels of antibodies able to react in immunoassays with the administered antigen [130]. A likely explanation of this difference is that B cell responses are degenerate and trigger the secretion not only of Igs that can function as detectable antibodies with the homologous antigen but also of heterospecific antibodies that have an affinity for the immunogen below the threshold required for binding to it although they may be able to bind to other, related antigens.
Specificity versus discrimination potential
The specificity of antibodies is inversely related to their ability to cross-react with a variety of antigens [131]. Part of the confusion surrounding the concept of specificity arises when no distinction is made between a single epitope and a multiepitopic antigen. If an investigator wishes to differentiate between two antigenically related proteins using a panel of Mabs, an antibody that recognizes the same epitope in both molecules would be called nonspecific whereas an antibody recognizing an epitope present in only one of them would be considered specific [93]. Since an antibody would thus be called specific or non specific, depending on what the investigator is trying to achieve, it is preferable to refer to the discrimination potential of antibodies raised against a multiepitopic antigen. An antibody, of course, cannot be specific for a multiepitopic protein and it will at best only have a limited specificity for an individual epitope. That an antibody is always able to cross-react with many related epitopes is also demonstrated by the ability of large numbers of peptide analogs presenting amino acid replacements at each residue position to react with the same antibody [50, 51, 132].
Antibody specificity and affinity
It is often believed that antibody specificity is correlated with high affinity because it is expected that highly specific antibodies will possess a better stereochemical complementarity with their antigens than antibodies of lower affinity. There is, however, no necessary link between affinity and specificity since antibodies of lower affinity may discriminate better between two antigens than do antibodies of higher affinity. The reason for this is that low affinity antibodies tend to detect fewer cross-reactions than antibodies of high affinity since cross-reactions are usually weaker than homologous reactions and will more quickly drop below the level of detection [93]. It must also be emphasized that in the same way that epitopes and paratopes are defined only through their mutual relationship, the affinity of an antibody can only be defined with respect to a binary relationship with a single antigen. In contrast, the specificity of an antibody is a ternary relational property that is meaningful only with respect to a more complex set of relations between one antibody and at least two antigens, the reason being that specificity arises from the ability of an antibody to react differently with two or more antigens and thus to discriminate between them.
Discontinuous epitopes cannot be extracted from their protein environment to be used as vaccine immunogens
The failure of reverse vaccinology in developing an HIV-1 vaccine is partly due to the unwarranted expectation that the structural delineation of discontinuous HIV-1 epitopes bound to neutralizing Mabs should allow these epitopes to be used as successful vaccine immunogens. As discussed above, a discontinuous epitope corresponds to a set of non-contiguous residues which cannot be isolated as a separate entity to show it possesses binding activity outside the protein context. Many unsuccessful attempts have been made to reconstitute the exact structure of discontinuous epitopes by chemical synthesis [78 and references therein; 133] and it does not seem possible to reproduce their exact immunological activity in the absence of the scaffolding contribution of the peptide chain. As a result, the immunogenic capacity of a discontinuous epitope can only be evaluated by using the native protein in which it is embedded as an immunogen, a procedure that will always stimulate an heterogeneous immune response against the numerous epitopes of the protein. For instance, when antibodies are raised against the CD4bs surface of gp120, most of them do not possess the exceptional neutralizing capacity of Mab b12 [134].
When a multiepitopic protein is used as immunogen, various epitopes will selectively bind to the B cell receptors present in the immunized host for which they possess sufficient affinity. However, the epitopes will not preferentially bind to those rare receptors, which in addition to recognizing the epitope, are responsible for the subsequent secretion by plasmocytes of antibodies that neutralize viral infectivity. The selection of B cell receptors during immunization is blind to the presence of neutralizing anti-viral activity in the antibodies that will be secreted since this biological function becomes effective only during the subsequent infection of a host. Furthermore, even if a single discontinuous epitope could somehow be used as an immunogen, the probability is still extremely low that a selectable B cell receptor would be present in the immunized host that exactly matches the variable immunoglobulin gene and CDR sequences of the Mab used for identifying the discontinuous epitope. Unfortunately, we do not know how to elicit neutralizing rather than non-neutralizing antibodies since it is not clear which features of an immunogen and immunized host are responsible for the cross-protective immunogenic activity that is required from any vaccine candidate [19, 135–137].
The unsuccessful search for immunogens able to elicit b12-like antibodies
The difficulties encountered when one attempts to design a vaccine immunogen on the basis of the known structure of a neutralizing antibody recognizing a discontinuous HIV-1 epitope are clearly demonstrated in the case of the b12 antibody. This Mab which was obtained from a phage-displayed Fab library derived from an HIV-1 patient [138] recognizes a conserved discontinuous epitope overlapping the CD4bs of gp120. It neutralizes a wide range of primary and TCLA HIV-1 isolates by inhibiting the binding of gp120 to the CD4 receptor. Antibody b12 has an 18-residue long CDR H3 region, a feature present in many anti-CD4bs human Mabs, allowing them to penetrate into a hydrophobic cavity on the gp120 surface. Using a docking model of b12 onto gp120, several contact residues of the epitope were identified and their contribution to binding activity was evaluated by alanine mutagenesis [26]. The gp120 structure that was used for docking b12 was the only one available at the time and corresponds to that of a monomeric core complexed with CD4 and with the Fab 17b [139], and it may therefore not correspond to the conformation of the trimeric CD4bs seen by antibodies and by the immune system. It is known that when gp120 binds to CD4, it undergoes a large conformational change which allows the virus to bind to one of its chemokine coreceptors [140, 141].
In an attempt to identify an HIV-1 vaccine immunogen capable of eliciting b12-like neutralizing antibodies, peptide libraries displayed on filamentous phage have been used to select peptides that bind b12 [84]. A 15-residue peptide labeled B2.1 with the sequence H1ERSYMFSDLENRCI15 was found to bind b12 with considerable affinity when it was present as a disulfide-bridged homodimer involving two C14 residues. The residues S8, D9, C14 and I15 of peptide B2.1 were present in several of the selected mimotope peptides and were considered to be responsible for most of their binding activity [84]. The peptide B2.1 shows significant homology to the D-loop region (residues: R273SVNFTDNAKTII285) of gp120 and appeared to be an exact conformational mimic of the D-loop region. When the immunogenicity of B2.1 was tested in mice and rabbits no antibodies were obtained that cross-reacted with gp120, indicating that B2.1 was not an effective candidate vaccine immunogen.
In another study, additional mimotopes reacting with b12 were obtained which possessed the same consensus sequence M/VθSD (where θ represents an aromatic amino acid) found in the D2.1 peptide [85]. Second and third generation libraries were constructed which gave rise to the improved consensus sequence G/SLL/MVWSDEL/H that reacted even more strongly with b12. Phage-displayed peptides containing this sequence competed with the binding of gp120 to b12 and were therefore considered to mimic the epitope recognized by b12. In contrast to what had been found with the B2.1 peptide, immunization with these more active phage-displayed mimotopes were able to elicit Env-specific antibodies, although immunization with the mimotopes conjugated to a carrier protein did not. Cross-reactive immune sera from immunized mice, however, did not neutralize laboratory-adapted strains of HIV-1 in vitro, showing that these improved mimotopes were also ineffective vaccine immunogens [85]. The conclusion that immunization with peptide mimics of discontinuous epitopes is not a feasible approach for developing epitope-targeted vaccines was corroborated in a recent study of mimotopes that mimicked discontinuous epitopes of three well-characterized proteins [142].
The inability of mimotopes reacting with the b12 antibody to elicit b12-like antibodies is due to the fact that these linear peptides do not reproduce the complex structural features of the discontinuous epitope recognized by Mab b12 and which presumably induced its formation. This was clearly demonstrated when the crystal structure of a complex between Fab of b12 and the B2.1 peptide was determined at 1.8 Å [143]. Only three of the critical binding residues of the B2.1 peptide (residues F7, S8 and D9) were found to form side-chain contacts with the b12 paratope while other residues important for the binding activity (Y5, C14, I15) only helped to stabilize the peptide structure. The B2.1 peptide is able to bind b12 not because it mimics the D-loop of gp120 but because it reacts with a different paratope subsite in b12 from the one that recognizes the complex discontinuous epitope determined by docking and mutagenesis studies [84, 124, 134]. Since the B2.1 peptide mimicks at best a very small portion of the discontinuous epitope recognized by b12, it is not surprising that this peptide is unable to elicit antibodies that cross-react with gp120 [143].
In an attempt to understand why Mab b12 possesses potent neutralizing properties whereas the majority of anti CDRbs Mabs do not, more than 60 residues of the CD4bs were mutated to alanine to determine whether these substitutions affected the ability of gp120 to bind to CD4 and to neutralizing and non-neutralizing Mabs [134]. Many substitutions were found to affect the binding of neutralizing and non-neutralizing Mabs in the same way, indicating a high degree of overlap in the epitopes recognized by these anti-CD4bs antibodies. Some of the mutations enhanced Mab binding but such an effect was never observed for CD4 binding, suggesting that the virus has evolved toward an optimal gp120-CD4 interaction. Although there were some individual differences between the effects of substitutions on the binding of different Mabs, no clear picture emerged that explained why b12 possessed a unique neutralizing capacity. Since certain residues in the V1/V2 and V3 loop of gp120 were able to affect the binding of b12, it was suggested that non-neutralizing anti-CD4bs Mabs might interact with the gp120 surface using a different orientation from the one used by b12 [134]. It is known that all anti-CD4bs Mabs bind monomeric gp120 similarly, independently of their neutralizing capacity, whereas only b12 is able to bind trimeric, functional gp120 presumably through neotopes present only on gp120 oligomers. This may be the reason why only native gp120 trimers are able to elicit an effective neutralizing antibody response [135, 144]. All attempts to find immunogens able to elicit b12-like antibodies were based on the premise that such immunogens must reproduce exactly the structure of the epitope recognized by b12 because it was assumed that the antigenic activity of an epitope is always accompanied by a related immunogenic activity. This assumption implies that antigenicity which is a chemical property arising from a molecular recognition process involving complementary antibodies is equivalent to immunogenicity which is a biological property leading to antibody secretion that occurs only in the context of an immunized host [145]. Immunogenicity depends on intrinsic properties of the immunogen but also on extrinsic factors such as the host Ig gene repertoire, the chance occurrence of appropriate B cell receptors, self-tolerance, an adequate production of cytokines, the presence of various cellular and regulatory mechanisms in the immunized host and the uncontrolled process of antibody affinity maturation [146, 147]. All these extrinsic factors did contribute to the occurrence of the neutralizing antibody initially used to identify the putative vaccine epitope but they cannot be expected to be present in identical form in the host that will be vaccinated with the same antigen. Another reason that makes it impossible to extrapolate from antigenic to immunogenic structures is that the structure of an epitope determined in a crystallographic complex is also influenced by processes of induced fit, conformational selection and mutual adaptation of the two binding partners [148–152]. As a result, the particular antigenic structure revealed in an antigen-antibody complex does not necessarily correspond to the immunogenic structure that was recognized by B cell receptors during the immunization process and should be present in the vaccine immunogen [29]. When tested in an immunoassay, a peptide may bind to an anti-protein neutralizing Mab by an induced fit mechanism driven by the pre-existing structure of the Mab. However, the same induced fit will not occur when the peptide is used as immunogen and is recognized by B cell receptors that have no special affinity for the cognate intact protein.
The assumption that immunogenic potential can be deduced from antigenic structures has led several groups to modify gp120 molecules in order to improve their vaccine potential. For instance, recombinant gp120s were generated that no longer were able to bind non-neutralizing anti CD4bs Mabs while retaining their capacity to bind b12, an approach called immunofocusing [153]. It was hoped that such a re-engineered gp120 would acquire the immunogenic capacity of eliciting only neutralizing antibodies, somehow mirroring the altered antigenic reactivity [134]. As was to be expected, there have been no subsequent reports indicating that the attempt to induce an immunogenic functionality in gp120 that mirrored the antigenic properties of the engineered constructs was successful. Recently a stabilized gp120 antigen, better able to bind neutralizing antibodies, has been developed by rational design [154]. The CD4bs of Env was re-engineered by deleting the V1 and V3 loops of the full gp120 molecule and introducing cross-links between different regions of the core protein. One of the re-engineered stabilized surfaces which reacted strongly with Mab b12 but not at all with four non-neutralizing CD4bs Mabs was used to select CD4bs-directed Mabs from memory B cells derived from a clade B-infected donor. A small number of CD4bs-specific memory B cells was obtained from about 25 millions peripheral blood mononuclear cells of the donor. One selected Mab (VRC01) neutralized about 90% of nearly 200 HIV isolates from multiple clades and had a much broader neutralizing capacity than b12 [154]. The authors suggested that their results do provide an important insight for future HIV-1 vaccine design, although they presented no evidence that their rationally designed antigen is also a superior immunogen able to induce VRC01-like polyclonal antibodies in humans.
Continuous epitopes of viral proteins are not effective vaccine immunogens
Although many linear peptide sequences in proteins cross-react with antibodies raised against the protein, they mostly are unable to elicit antibodies that cross-react with the native protein. The lack of cross-reactive immunogenicity of peptides [136] can be explained as follows. When an antiprotein antibody is tested for it ability to react with a peptide in an immunoassay, it may select one of the many available peptide conformations or it may bring it about by induced fit, the result in both cases being a detectable cross-reaction. In contrast, when the same peptide is confronted with a variety of B cell receptors during immunization, different conformations of the peptide may be recognized by separate receptors, leading to the subsequent appearance of a variety of antipeptide antibodies. However, there is no reason why the peptide would bind preferentially to those rare receptors which in addition also cross-react with a more complex epitope present only in the native protein. Since a peptide corresponding to a continuous epitope is nearly always a poor mimic of a protein discontinuous epitope, it is unlikely that a significant proportion of the elicited antipeptide antibodies would react with the cognate protein. Antibodies which do not recognize viral proteins will, of course, possess no neutralizing activity. It is sometimes possible to improve the cross-reactive immunogenicity of a peptide by increasing the conformational similarity between peptide and intact protein, for instance by cyclization of the peptide, but this approach rarely succeeds in producing immunogens able to elicit neutralizing antibodies [78 and references therein; 155, 156]. As discussed earlier in the case of discontinuous epitopes, linear peptides corresponding to continuous epitopes, when used as immunogens, will not preferentially select and bind to the rare B cell receptors which in addition to cross-reacting with the native protein also trigger a B cell differentiation process giving rise to antibodies endowed with a functional protective activity. Although a few mimotopes of several continuous epitopes of different viruses, including HIV-1, have been found to be able to elicit antibodies that cross-react with the virus and possess some neutralizing activity [75, 83, 88], no successful protective peptide-based antiviral vaccine has been developed and marketed for human use [157]. Two 6-residue long continuous epitopes located in the conserved region of MPER of gp41 have been regarded as promising HIV-1 vaccine candidates [18] and many groups have attempted to turn them into effective vaccine immunogens [5]. It seems highly unlikely, however, that these short linear peptides which are part of larger complex discontinuous epitopes eliciting potent neutralizing Mabs, will be able to induce an effective protective immune response [29].
The special case of terminal regions in proteins
The terminal regions of proteins frequently harbour continuous epitopes because they tend to be surface-oriented [158] and are more hydrophilic and mobile than internal regions [158–161]. A major purpose of epitope prediction is to replace intact antigen molecules by linear synthetic peptides for use as diagnostic reagents for detecting viral antibodies [162] or as immunogens for raising antipeptide antibodies able to cross-react with a parent protein [163, 164]. The most effective strategy in this case is to select 10-residue long synthetic peptides corresponding to the N- and C- terminal regions of the protein since these have the highest probability of cross-reacting antigenically with the protein. Methods for predicting which continuous epitopes in proteins are likely to be effective vaccine immunogens have been notoriously unsuccessful [43, 44, 165, 166] and only very limited success has been obtained when certain terminal peptides were used as vaccine immunogens. Vaccination with the N-terminal 15-residue peptide of the VP2 protein of canine parvovirus, for instance, was able to protect dogs against virus infection [167]. Another example is the C-terminal region of the VP1 protein (residues 200-213) of foot-and-mouth disease virus, which is able to elicit high levels of neutralizing antibodies [168]. Residues 200-213 of one VP1 subunit are located next to the immunodominant disordered loop (residues 141-160) of an adjacent subunit [169], and both regions contribute to a single discontinuous neotope [170]. The ability of the terminal regions of proteins to induce antibodies that cross-react with the native protein is partly due to the fact that these regions possess an intrinsic disorder which makes them very similar to the unstructured peptides [171]. Although some disordered regions in proteins may be recognized preferentially by antipeptide antibodies, the majority of viral epitopes capable of inducing a neutralizing antibody response are actually complex, discontinuous topographic sites rather than terminal linear peptides.
The HIV-1 V3 antigenic site
Loops and turns in proteins are often immunodominant antigenic regions and the V3 loop in HIV-1 has for many years been considered the principal neutralizing domain of the virus [10, 172]. Although it is designated as the third variable domain of gp120, the V3 loop has many conserved features such as a constant size of about 35 residues, a conserved type II turn at its tip and a disulfide bond at its base [173]. The semiconserved nature of the V3 loop allows it to play an important role in the recognition of viral co-receptors, but its sequence variability has led investigators to question its value as a target for vaccine-induced antibodies. Another reason that led investigators to doubt the value of the V3 loop as a vaccine candidate is that they looked for the presence of anti V3 antibodies in human anti-HIV-1 antisera by absorbing the sera with linear V3 peptides [174]. Since this absorption step did not remove the antibodies that neutralized primary HIV-1 isolates, it was concluded that the neutralizing antibodies were not directed against the V3 loop. In fact, human sera that neutralize both TCLA isolates and primary isolates from several HIV-1 clades contain antibodies that recognize a cyclic V3 conformation present in the virus and in disulfide-bonded V3 loops but absent in linear V3 peptides [4, 14, 17, 175, 176]. The V3 loop is very flexible and is able to adopt different conformations when it binds to different Mabs [177]. It was found that alternative β-hairpin conformations can be induced in the same V3 peptide, depending on whether it binds to Mab 447-52D that neutralizes both R5 and X4 viruses or to Mab 5β which neutralizes only X4 viruses [178]. This shows that the epitope conformation observed in a Mab-epitope complex can result from an induced fit process and is therefore not a reliable guide for deciding which immunogen conformation should be used for eliciting a certain type of neutralizing antibody. It is not yet clear whether intrinsically flexible V3 loops should be constrained in a particular conformation to increase their potential value as vaccine candidates [179]. In the case of the immunodominant GH loop (residues 141-160) of the VP1 protein of foot-and-mouth disease virus, a stabilized all-D retro loop synthesized with D-amino acids was found to induce higher levels of neutralizing antibodies that the corresponding L-peptide [180–182].
One of the many hurdles to be overcome by an HIV-1 vaccine is the antigenic variability of the virus
There are several reasons why the strategies that were used successfully in the past to develop effective vaccines against many viral diseases are not applicable to HIV-1 [183–185]: (1) Since the natural immune response in HIV-1 infected individuals does not clear the infection, there is no natural immunological mechanism that the vaccine could imitate. An HIV-1 vaccine would thus have to surpass the normal immune response induced by infection and would for instance have to generate a far stronger response against conserved epitopes than what occurs during infection; (2) HIV-1 integrates into the host genome and establishes a latent pool of infected cells that conceal the virus from immune recognition; (3) The virus progressively destroys the immune system; (4) HIV-1 isolates exhibit an enormous antigenic variability arising from the error-prone viral reverse transcription which can produce as much as 35% sequence diversity in gp120 between viral subtypes and 10% diversity in gp120 in a single infected individual.
Although an increasing number of broadly neutralizing Mabs have recently been isolated from infected human donors [22, 186–189], such antibodies are rarely induced by experimental immunization. It seems that during HIV-1 infection, the immune response is mostly directed against highly variable and accessible Env regions such as the V1, V2 and V3 loops rather than against functionally important but less accessible conserved domains such as the receptor and co-receptor binding sites. The propensity of the immune system to respond to immunodominant, strain-specific highly variable antigenic sites instead of to subdominant conserved epitopes that are able in principle to elicit a more broadly protective immunity, has been called deceptive imprinting because it allows the virus to escape immune pressure [190]. Attempts to overcome deceptive imprinting by redirecting the immune response to less strain-specific epitopes have been made using a strategy called immune refocusing. By introducing several new glycosylation sites into the V1 and V3 loops, it was possible for instance to obtain immunogens that induced a broader neutralization response that was effective against heterologous strains [191]. Some of the reported immunization results, however, are difficult to interpret because linear instead of cyclized loop peptides were used in inhibition experiments aimed at determining the specificity of the induced antibodies. Attempts have also been made to redirect the immune response to conserved neutralization epitopes by deleting some or most of the variable loops from Env, but the results were not satisfactory [192, 193]. Other approaches to overcome the problem of extensive antigenic variability include using as immunogen either artificial consensus sequences based on the most common amino acid in each position in an alignment [194, 195], or alternatively combinatorial convergent peptide libraries consisting of thousands of V3 peptides known as mixotopes that are recognized as a single entity by immune receptors [196]. Other strategies to deal with the existence of many antigenic variants by means of polyvalent vaccine cocktails have also been proposed [197]. Recent results have shown that the V3 loop may, in fact, be able to induce a neutralizing response against many different HIV-1 clades, indicating that a polyclonal response to V3 may have a much greater neutralizing breadth than that displayed by any single Mab or even a cocktail of Mabs [17, 173, 198].
Rational design versus empirical immunogenicity trials in HIV-1 vaccine development
A pervading theme in current vaccine research is the view that rational design offers the best prospects for the much needed development of effective vaccines against HIV-AIDS, tuberculosis and malaria [199–202]. In the case of HIV-1, this viewpoint has led to the claim that “rational design represents the only approach that can elevate vaccine research from an empirical exercise to a scientific discipline” [27]. It is argued here that such a view which denigrates the time-honoured empirical, trial-and-error approach that gave us all our present day effective vaccines is highly misleading and could even possibly jeopardize future efforts at developing an HIV-1 vaccine [107, 109].
It is not always clear what scientists mean when they refer to rational design and it may be helpful to briefly analyze these terms. The label rational tends to be used to describe any approach that relies on the probable outcome of an intervention as predicted from available scientific knowledge. The term rational is also used to describe common sense decisions such as focusing on known antigenic regions of a viral protein as primary targets for a vaccine [203]. Since all scientific work is based on reason, logic and currently accepted scientific theories, it makes little sense to oppose rational and empirical approaches since all scientific knowledge is derived from empirical observations made in the course of experiments that are necessarily planned and analyzed in a rational manner. It is therefore misleading to imply that trial-and-error experimentation is not rational since all novel findings and discoveries arise from the unpredictable outcome of controlled experiments rather than from purely rational, deductive thinking [107]. The term design can be defined as the deliberate and intelligent conceiving of an artificial object or process. The designer’s task is to pose and solve an inverse problem, namely to imagine on the basis of existing knowledge what would bring about a desired outcome [204]. Plausible solutions can only be tested by trial and error until the preset goal is attained [205]. Since scientists need to secure funding for their research, it is tempting for them to suggest that their current understanding of the complexity of the immune system is sufficient to allow them to design immunogens and to predict the type of antibodies they will elicit.
Rational design terminology implies that there is a causal relationship between the structure of an antibody and its function, giving rise to the assertion: “structure determines function”. Although it is true that a biological activity always depends on an underlying structure, the structure does not possess causal efficacy on its own in bringing about a certain activity. Causal relations are relations between successive events and not between two material objects nor between a structure and an event [206]. There is no unique causal relation between the structure of an antibody and its neutralizing capacity since the occurrence of neutralization is influenced by a large number of immunological and pathophysiological factors arising from complex interactions between antibody, pathogen, and host. The isolation of the potent neutralizing Mab VRC01 [154] and the elucidation of its structure [207], as well as the isolation of many additional broadly neutralizing antibodies from the serum of infected donors [187–189, 208, 209], has been interpreted as providing valuable molecular information that should facilitate rational vaccine design [210]. The search for additional broadly neutralizing Mabs has been stimulated by the finding that it is possible to provide sterilizing protection in non-human primates by administering neutralizing Mabs prior to viral challenge. However, this type of successful immunotherapy using neutralizing antibodies derived from infected individuals does not in any way tell us how such antibodies can be induced by vaccination. It is precisely our lack of knowledge regarding which immunogens are able to induce such antibodies in certain infected individuals which prevents us from developing an HIV-1 vaccine by design. Furthermore, the polyspecificity of antibody molecules and the degeneracy of the immune system imply that it is futile to search for the single “intrinsic” epitope of a neutralizing Mab and for the putative unique immunogenic structure capable of inducing protective antibodies. It has been claimed [185] that our failure to develop an HIV-1 vaccine is due to the fact that we do not understand what type of immune response is needed for protection. We can of course surmise that effective CD4, CD8 and B-cell responses are needed but this is of little help since we do not know how to elicit the required responses by immunization. One could also argue that studying the mechanism of a preventive or therapeutic HIV-1 vaccine will become possible only once an effective vaccination protocol has been discovered empirically. Figure 2 lists many of the questions that we need to answer if we want to be able to rationally design an HIV-1 vaccine [109, 185, 211]. It has been suggested that rational design may not be the best strategy to follow [212] since it is not possible, for instance, to predict which combination of several substitutions in a protein may lead to new desired properties that are not manifested when the protein presents only one substitution. This has led to the conclusion that an improved protein function is more likely to be obtained by successive rounds of random mutation, recombination and selection than by rational design [206 and references therein]. What is of course possible is to rationally design an antigen to fit a single Mab [154, 213] or to improve by molecular design the paratope binding efficacy of a Mab intended for passive immunotherapy. What is not possible is to rationally design an immunogenic molecule that will elicit a protective polyclonal antibody response of predetermined efficacy.

Unanswered questions regarding the factors and mechanisms believed to be important for inducing an effective HIV-1 vaccine immune response [185], reproduced with permission
The existence among HIV-1 subjects of long-term non-progressors (LTNPs) and elite controllers (ECs) has given rise to the hope that the elucidation of the mechanisms responsible for their occurrence may help HIV-1 vaccine design [214]. LTNPs which make up between 2 and 5% of all HIV-1 seropositive individuals [215] maintain low plasma levels of HIV-RNA and elevated CD4+ T cell levels whereas the even rarer EC individuals have undetectable levels of viral RNA. A combination of viral, genetic and immunological factors has been implicated in the ability of these slow progressors to control HIV-1 infection. The principal viral factor believed to slow disease progression is HIV-1 strain attenuation arising from mutations in viral accessory genes that cause a reduction in virus infectivity and replication [214]. If control of disease progression in these individuals results from an infection with attenuated, poorly replicating viruses, it is not clear how this could be made use of in vaccine development. The same applies to host genetic and immunological factors that promote control of infection such as chemokine receptor polymorphisms or the presence of certain protective HLA class I alleles in LTNP and EC individuals. Although some slow progressors have been shown to be able to induce broadly neutralizing antibodies against multiple HIV-1 strains [213, 216], the relevance of this anti-Env immune response to vaccine design is also not obvious since we do not know which immunogens were able to induce such as a response. It is, indeed, generally recognized that the major bottleneck in designing an HIV-1 vaccine lies in our ignorance of which immunogens are capable of inducing a broadly neutralizing immune response [22, 189, 201]. As discussed in this review, a successful design strategy requires that we should know which immunogens and immunological mechanisms are likely to lead to the appearance of protective immunity following vaccination. Unfortunately even a vaccine that induces high levels of neutralizing antibodies will not necessarily protect against virus infection [217]. Many investigators have recently stressed the need for embarking on large scale research programs in basic and preclinical immunology which they believe will give us the understanding required for designing vaccines [185, 201]. Although nobody would quarrel with the need for more basic research in immunology, the track record in vaccinology shows that effective vaccines in the past were always discovered by trial-and-error immunization trials rather than by rational design based on available structural and immunological knowledge. The widespread current expectation that discoveries in biomedicine and vaccinology are likely to result from structure-based molecular studies, “omics” type investigations [202] or systems biology approaches [218] rather than from empirical trials may well turn out to be counterproductive if it inhibits investigators from undertaking exploratory trial-and-error experiments. Empirical immunogenicity trials based on a less than perfect understanding of the complexities of the immune response and of the nature of dynamic and transitory HIV-1 epitopes may still be the key to discovering a promising HIV-1 vaccine candidate. Although epitope mapping with neutralizing Mabs and structural analyses may help investigators to identify which regions of the viral surface should be targeted by a vaccine, it must be emphasized that antigen mapping on its own will not lead to the discovery of vaccine immunogens that can only be revealed following experimental immunogenicity trials. Identifying the many cross-reactive epitopes that a neutralizing Mab can recognize is an entirely different task from discovering empirically which vaccine immunogens are able to induce a protective immune response.
Once an effective immunogen has been identified empirically, it may still take many years of basic research to establish its mechanism of action. As is the case with most present day successful vaccines, it is even conceivable that we may never fully understand why an empirically discovered vaccine is actually effective. This is a salutary reminder that discovering an effective vaccine empirically should have a higher priority than elucidating the mode of action of an ideal HIV-1 vaccine. Basic immunological research, therefore, should not stand in the way of rational immunogenicity trials nor should it restrict the financial resources that must be made available to implement them.
Conclusions
Creative imaginative thinking is required to choose credible candidate vaccines and to invent plausible hypotheses to help select judicious vaccine regimens, dosages, adjuvants, delivery routes and effective T cell help. Instead of misrepresenting experimental hypothesis testing as a case of rational design, it would seem preferable to openly acknowledge that the empirical testing of immunogens, which initially should take the form of small human trials, still offers the best hope of discovering an effective HIV-1 vaccine. There is increasing doubt that developing a preventive vaccine able to provide prophylactic immunity to stop HIV-1 infection occurring altogether is a realistic enterprise. There is indeed no evidence that it is possible to induce sufficiently high levels of broadly neutralizing antibodies at mucosal surfaces to achieve that aim. In recent years, much research has been directed instead to the development of therapeutic HIV-1 vaccines for treating people who are already infected with HIV-1. It has been shown, for instance, that immunization with the conserved HIV regulatory proteins, Tat, Rev and Nef, which are the first proteins expressed after HIV infection, is able to stimulate the immune system of patients receiving antiretroviral therapy, enabling them to partially control virus replication [30]. Other therapeutic vaccines are currently being investigated [219, 220] and it may become possible to assess their value more rapidly in the future by using so-called «adaptive trial designs» aimed at screening out poor vaccines while extending the evaluation of efficacious ones [221]. In addition, since many methods are now available to chemically inactivate HIV-1 while retaining the conformational and functional integrity of the gp proteins [29 and references therein], it may be justified to re-examine the possible value of a killed HIV-1 vaccine, provided the total innocuity of inactivated and genetically modified virus can be established.
The unexpected promising results of the Thailand “RV144” trial, initially condemned as ill-conceived and unjustified [222–225] but which turned out to be the first human vaccine trial showing modest protection against HIV infection [226, 227], demonstrate that empirical trials will always be needed since their outcomes are inherently unpredictable.
References
Klatt NR, Chahroudi A, Silvestri G (2010) Human immunodeficiency viruses: pathogenesis. In: Mahy BWJ, Van Regenmortel MHV (eds) Desk encyclopedia of human and medical virology. Academic Press, Elsevier, Oxford, pp 345–353
Norrby E (ed) (1993) Immunochemistry of AIDS. Karger, Switzerland
Wyatt R, Kwong PD, Desjardins E et al (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393:705–711
Zolla-Pazner S (2004) Identifying epitopes of HIV-1 that induce protective antibodies. Nat Rev Immunol 4:199–210
Montero M, van Houten NE, Wang X, Scott JK (2008) The membrane proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol Mol Biol Rev 72:54–84
Hoxie JA (2010) Toward an antibody-based HIV-1. Vaccine Annu Rev Med 61:135–152
Poignard P, Moulard M, Golez E et al (2003) Heterogeneity of envelope molecules expressed on primary human immunodeficiency virus type 1 particles as probed by the binding of neutralizing and non neutralizing antibodies. J Virol 77:353–365
Moore PL, Crooks ET, Porter L et al (2006) Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J Virol 80:2515–2528
Crooks ET, Moore PL, Franti M et al (2007) A comparative immunogenicity study of HIV-1 virus-like particles bearing various forms of envelope proteins, particles bearing no envelope and soluble monomeric gp120. Virology 366:245–262
Javaherian K, Langlois AJ, McDanal C et al (1989) Principal neutralizing domain of the human immunodeficiency virus type 1 envelope protein. Proc Natl Acad Sci USA 86:6768–6772
Muster T, Steindl F, Purtscher M et al (1993) A conserved neutralizing epitope on gp41 of human immunodeficiency virus type 1. J Virol 67:6642–6647
Zwick MB, Labrijn AF, Wang M et al (2001) Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J Virol 75:10892–10905
Poignard P, Saphire EO, Parren PW, Burton DR (2001) gp120: biologic aspects of structural features. Ann Rev Immunol 19:253–274
Gorny MK, Williams C, Volsky B et al (2002) Human monoclonal antibodies specific for conformation-sensitive epitopes of V3 neutralize human immunodeficiency virus type 1 primary isolates from various clades. J Virol 76:9035–9045
Dhillon AK, Donners H, Pantophlet R et al (2007) Dissecting the neutralizing antibody specificities of broadly neutralizing sera from human immunodeficiency virus type 1-infected donors. J Virol 81:6548–6562
Li Y, Migueles SA, Welcher B et al (2007) Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat Med 13:1032–1034
Hioe CE, Wrin T, Seaman MS et al (2010) Anti-V3 monoclonal antibodies display broad neutralizing activities against multiple HIV-1 subtypes. PLoS One 5:e10254
Zwick MB (2005) The membrane-proximal external region of HIV-1 gp41: a vaccine target worth exploring. AIDS 19:1725–1737
Zwick MB, Burton DR (2007) HIV-1 neutralization: mechanisms and relevance to vaccine design. Curr HIV Res 5:608–624
Willey S, Aasa-Chapman MM (2008) Humoral immunity to HIV-1: neutralization and antibody effector functions. Trends Microbiol 16:596–604
Binley J (2009) Specificities of broadly neutralizing anti-HIV-1 sera. Curr Opin HIV AIDS 4:364–372
Stamatatos L, Morris L, Burton DR, Mascola JR (2009) Neutralizing antibodies generated during natural HIV-1 infection: good news for an HIV-1 vaccine? Nat Med 15:866–870
Burton DR (2002) Antibodies, viruses and vaccines. Nat Rev Immunol 2:706–713
Burton DR (2010) Scaffolding to build a rational vaccine design strategy. Proc Natl Acad Sci USA 107:17859–17860
Schief WR, Ban YE, Stamatatos L (2009) Challenges for structure-based HIV vaccine design. Curr Opin HIV AIDS 4:431–440
Saphire EO, Parren PWHI, Pantophlet R et al (2001) Crystal structure of a neutralizing human IgG against HIV-1: a template for vaccine design. Science 293:1155–1159
Karlsson-Hedestam GB, Fouchier RA, Phogat S et al (2008) The challenges of eliciting neutralizing antibodies to HIV-1 and to influenza virus. Nat Rev Microbiol 6:143–155
Van Regenmortel MHV (2011) Two meanings of reverse vaccinology and the empirical nature of vaccine science. Vaccine 29:7875
Van Regenmortel MHV (2011) Limitations to the structure-based design of HIV-1 vaccine immunogens. J Mol Recognit 24:741–753
Caputo A, Gavioli R, Bellino S et al (2009) HIV-1 Tat-based vaccines: an overview and perspectives in the field of HIV/AIDS vaccine development. Int Rev Immunol 28:285–334
Atassi MZ, Smith JA (1978) A proposal for the nomenclature of antigenic sites in peptides and proteins. Immunochemistry 15:609–610
Barlow DJ, Edwards MS, Thornton JM (1986) Continuous and discontinuous protein antigenic determinants. Nature 322:747–748
Sundberg EJ, Mariuzza RA (2002) Molecular recognition in antibody-antigen complexes. Adv Protein Chem 61:119–160
Thali M, Olshevsky U, Furman C et al (1991) Characterization of a discontinuous human immunodeficiency virus type 1 gp120 epitope recognized by a broadly reactive neutralizing human monoclonal antibody. J Virol 65:6188–6193
Moore JP, Ho DD (1993) Antibodies to discontinuous or conformationally sensitive epitopes on the gp120 glycoprotein of human immunodeficiency virus type 1 are highly prevalent in sera of infected humans. J Virol 67:863–875
VanCott TC, Bethke FR, Burke DS et al (1995) Lack of induction of antibodies specific for conserved, discontinuous epitopes of HIV-1 envelope glycoprotein by candidate AIDS vaccines. J Immunol 155:4100–4110
Zwick MB, Kelleher R, Jensen R et al (2003) A novel human antibody against human immunodeficiency virus type 1 gp120 is V1, V2, and V3 loop-dependent and helps delimit the epitope of the broadly neutralizing antibody immunoglobulin G1 b12. J Virol 77:6965–6978
Laver WG, Air GM, Webster RG, Smith-Gill SJ (1990) Epitopes on protein antigens: misconceptions and realities. Cell 61:553–556
Van Regenmortel MHV (2009) What is a B cell epitope? In: Reineke U, Schutkowski M (eds) Epitope mapping protocols. Humana Press, Springer, pp 3–20
Hopp TP (1993) Retrospective: 12 years of antigenic determinant predictions, and more. Pept Res 6:183–190
Hopp TP (1994) Different views of protein antigenicity. Pept Res 7:229–231
Van Regenmortel MHV, Pellequer J-L (1994) Predicting antigenic determinants in proteins: looking for unidimensional solutions to a three-dimensional problem? Pept Res 7:224–228
Ponomarenko JV, Van Regenmortel MHV (2009) B cell epitope prediction. In: Gu J, Bourne PE (eds) Structural bioinformatics, 2nd edn. John Wiley, Hoboken, pp 849–879
Greenbaum JA, Andersen PH, Blythe M et al (2007) Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J Mol Recognit 20:75–82
Zhang Q, Wang P, Kim Y, Haste-Andersen P et al (2008) Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res 36:W513–W518
Van Regenmortel MHV (1999) Molecular dissection of protein antigens and the prediction of epitopes. In: Van Regenmortel MHV, Muller S (eds) Synthetic peptides as antigens. Elsevier, Amsterdam, pp 1–78
Chen S-WW, Van Regenmortel MHV, Pellequer J-L (2009) Structure-activity relationships in peptide-antibody complexes: implications for epitope prediction and development of synthetic peptide vaccines. Curr Med Chem 16:953–964
Muller S, Plaué S, Couppez M, Van Regenmortel MHV (1986) Comparison of different methods for localizing antigenic regions in histone H2A. Mol Immunol 23:593–601
Trifilieff E, Dubs MC, Van Regenmortel MHV (1991) Antigenic cross-reactivity potential of synthetic peptides immobilized on polyethylene rods. Mol Immunol 28:889–896
Getzoff ED, Tainer JA, Lerner RA, Geysen HM (1988) The chemistry and mechanism of antibody binding to protein antigens. Adv Immunol 43:1–98
Geysen HM, Mason TJ, Rodda SJ (1988) Cognitive features of continuous antigenic determinants. J Mol Recognit 1:32–41
Jemmerson R (1987) Antigenicity and native structure of globular proteins: low frequency of peptide reactive antibodies. Proc Natl Acad Sci USA 84:9180–9184
Spangler BD (1991) Binding to native proteins by antipeptide monoclonal antibodies. J Immunol 146:1591–1595
Lerner RA (1984) Antibodies of predetermined specificity in biology and medicine. Adv Immunol 36:1–44
Darst SA, Robertson CR, Berzofsky JA (1988) Adsorption of the protein antigen myoglobin affects the binding of conformation-specific monoclonal antibodies. Biophys J 53:533–539
Hafenstein S, Bowman VD, Sun T et al (2009) Structural comparison of different antibodies interacting with parvovirus capsids. J Virol 83:5556–5566
Van Regenmortel MHV (1966) Plant virus serology. Adv Virus Res 12:207–271
Van Regenmortel MHV (1992) The conformational specificity of viral epitopes. FEMS Microbiol Lett 100:483–487
Neurath AR, Rubin BA (1971) Viral structural components as immunogens of prophylactic value. Monographs in virology. Karger, Basel
Van Regenmortel MHV, Neurath AR (1985) Immunochemistry of viruses. Elsevier, Amsterdam
Broder CC, Earl PL, Long D et al (1994) Antigenic implications of human immunodeficiency virus type 1 envelope quaternary structure: oligomer-specific and -sensitive monoclonal antibodies. Proc Natl Acad Sci USA 91:11699–11703
Cho MW, Lee MK, Chen CH et al (2000) Identification of gp120 regions targeted by a highly potent neutralizing antiserum elicited in a chimpanzee inoculated with a primary human immunodeficiency virus type 1 isolate. J Virol 74:9749–9754
Gorny MK, Stamatatos L, Volsky B et al (2005) Identification of a new quaternary neutralizing epitope on human immunodeficiency virus type 1 virus particles. J Virol 79:5232–5237
Bothner B, Dong XF, Bibbs L et al (1998) Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J Biol Chem 9:673–676
Yuan W, Bazick J, Sodroski J (2006) Characterization of the multiple conformational states of free monomeric and trimeric human immunodeficiency virus envelope glycoproteins after fixation by cross-linker. J Virol 80:6725–6737
Jerne NK (1960) Immunological speculations. Annu Rev Microbiol 14:341–358
Bou-Habib DC, Roderiquez G, Oravecz T et al (1994) Cryptic nature of envelope V3 region epitopes protects primary monocytotropic human immunodeficiency virus type 1 from antibody neutralization. J Virol 68:6006–6013
Poignard P, Klasse PJ, Sattentau QJ (1996) Antibody neutralization of HIV-1. Immunol Today 17:239–246
Geysen HM, Rodda SJ, Mason TJ (1986) A priori delineation of a peptide which mimics a discontinuous antigenic determinant. Mol Immunol 23:709–715
Irving MB, Pan O, Scott JK (2001) Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics. Curr Opin Chem Biol 5:314–324
Hanin V, Déry O, Boquet D et al (1995) Importance of hydropathic complementarity for the binding of the neuropeptide substance P to a monoclonal antibody: equilibrium and kinetic studies. Mol Immunol 34:829–838
Van Regenmortel MHV (1998) Mimotopes, continuous paratopes and hydropathic complementarity: novel approximations in the description of immunological specificity. J Disper Sci Technol 8:140–144
Blalok J (1990) Complementarity of peptides specified by “sense” and “antisense” strands of DNA. Trends Biotechnol 8:140–144
Tropsha A, KizlerJS Chaiken IM (1992) Making sense of antisense: a review of experimental data and developing ideas on sense-antisense recognition. J Mol Recognit 5:43–54
Yu MW, Scott JK, Fournier A, Talbot PJ (2000) Characterization of murine coronavirus neutralization epitopes with phage-displayed peptides. Virology 271:182–196
Malby RL, Tulip WR, Harley VR et al (1994) The structure of a complex between the NC10 antibody and influenza virus neuraminidase and comparison with the overlapping binding site of the NC41 antibody. Structure 2:733–746
Lescar J, Pellegrini M, Souchon H et al (1995) Crystal structure of a cross-reaction complex between Fab F9.13.7 and guinea fowl lysozyme. J Biol Chem 270:18067–18076
Van Regenmortel MHV (2009) Synthetic peptide vaccines and the search for neutralization B cell epitopes. Open Vaccine J 2:33–44
Delmastro P, Meola A, Monaci P et al (1997) Immunogenicity of filamentous phage displaying peptide mimotopes after oral administration. Vaccine 15:1276–1285
Puntoriero G, Meola A, Lahm A et al (1998) Towards a solution for hepatitis C virus hypervariability: mimotopes of the hypervariable region 1 can induce antibodies cross-reacting with a large number of viral variants. EMBO J 17:3521–3533
Larralde OG, Martinez R, Camacho F et al (2007) Identification of hepatitis A virus mimotopes by phage display, antigenicity and immunogenicity. J Virol Methods 140:49–58
Schellekens GA, Lasonder E, Feijlbrief M et al (1994) Identification of the core residues of the epitope of a monoclonal antibody raised against glycoprotein D of herpes simplex virus type 1 by screening of a random peptide library. Eur J Immunol 24:3188–3193
Steward MW, Stanley CM, Obeid OE (1995) A mimotope from a solid-phase peptide library induces a measles virus-neutralizing and protective antibody response. J Virol 69:7668–7673
Zwick MB, Bonnycastle LL, Menendez A et al (2001) Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J Virol 75:6692–6699
Dorgham K, Dogan I, Bitton N et al (2005) Immunogenicity of HIV type 1 gp120 CD4 binding site phage mimotopes. AIDS Res Hum Retrovir 21:82–92
Keller PM, Arnold BA, Shaw AR et al (1993) Identification of HIV vaccine candidate peptides by screening random phage epitope libraries. Virology 193:709–716
Boots LJ, McKenna PM, Arnold BA et al (1997) Anti-human immunodeficiency virus type 1 human monoclonal antibodies that bind discontinuous epitopes in the viral glycoproteins can identify mimotopes from recombinant phage peptide display libraries. AIDS Res Hum Retrovir 13:1549–1559
Scala G, Chen X, Liu W et al (1999) Selection of HIV-specific immunogenic epitopes by screening random peptide libraries with HIV-1-positive sera. J Immunol 162:6155–6161
Cohn M (1997) A new concept of immune specificity emerges from a consideration of the self-nonself discrimination. Cell Immunol 181:103–108
Cohn M (2005) Degeneracy, mimicry and cross-reactivity in immune recognition. Mol Immunol 42:651–655
Langman RE (2000) The specificity of immunological reactions. Mol Immunol 37:555–561
Cohen IR, Hershberg U, Solomon S (2004) Antigen-receptor degeneracy and immunological paradigms. Mol Immunol 40:993–996
Van Regenmortel MHV (1998) From absolute to exquisite specificity. Reflections on the fuzzy nature of species, specificity and antigenic sites. J Immunol Methods 216:37–48
Mazumdar PH (1995) Species and specificity. Cambridge University Press, Cambridge
Landsteiner K (1947) The specificity of serological reactions. Harvard University Press, Cambridge
Talmadge DW (1959) Immunological specificity, unique combinations of selected natural globulins provide an alternative to the classical concept. Science 129:1643–1648
Medawar PB, Medawar JS (1978) The life science. Granada Publishing, London
Bhattacharjee AK, Glaudemans CP (1978) Dual binding specificities in MOPC 384 and 870 murine myeloma immunoglobulins. J Immunol 120:411–413
Ramsland PA, Guddat LW, Edmundson AB, Raison RL (1997) Diverse binding site structures revealed in homology models of polyreactive immunoglobulins. J Comput Aided Mol Des 11:453–461
James LC, Roversi P, Tawfik DS (2003) Antibody multi-specificity mediated by conformational diversity. Science 299:1362–1367
Jimenez R, Salazar G, Baldridge KK, Romesberg FE (2003) Flexibility and molecular recognition in the immune system. Proc Natl Acad Sci USA 100:92–97
Sperling R, Francus T, Siskind GW (1983) Degeneracy of antibody specificity. J Immunol 131:882–885
Parnes O (2004) From interception to incorporation: degeneracy and promiscuous recognition as precursors of a paradigm shift in immunology. Mol Immunol 40:985–991
Wucherpfennig KW, Allen PM, Celada F et al (2007) Polyspecificity of T cell and B cell receptor recognition. Semin Immunol 19:216–224
Edelman GM, Gally JA (2001) Degeneracy and complexity in biological systems. PNAS 98:13763–13768
Moodie SL, Mitchell JB, Thornton JM (1996) Protein recognition of adenylate: an example of a fuzzy recognition template. J Mol Biol 263:486–500
Van Regenmortel MHV (1999) Molecular design versus empirical discovery in peptide based vaccines. Coming to terms with fuzzy recognition sites and ill-defined structure-function relationships in immunology. Vaccine 18:216–221
McNeil D, Freiberger P (1993) Fuzzy logic. Simon and Schuster, New-York
Van Regenmortel MHV (2001) Pitfalls of reductionism in the design of peptide-based vaccines. Vaccine 19:2369–2374
Sadegh-Zadeh K (2001) The fuzzy revolution: goodbye to the Aristotelian Weltanschauung. Artific Intell Med 21:1–25
Leng Q, Bentwich Z (2002) Beyond self and nonself: fuzzy recognition of the immune system. Scand J Immunol 56:224–232
Efroni S, Cohen IR (2002) Simplicity belies a complex system: a response to the minimal model of immunity of Langman and Cohn. Cell Immunol 216:23–30
Sandberg JK, Franksson L, Sundbäck J et al (2000) T cell tolerance based on avidity thresholds rather than complete deletion allows maintenance of maximal repertoire diversity. J Immunol 165:25–33
Foote J, Eisen HN (1995) Kinetic and affinity limits on antibodies produced during immune responses. Proc Natl Acad Sci USA 92:1254–1256
Batista FD, Neuberger MS (1998) Affinity dependence of the B cell response to antigen: a threshold, a ceiling, and the importance of off-rate. Immunity 8:751–759
Schroer JA, Bender T, Feldmann T, Kim KJ (1983) Mapping epitopes on the insulin molecule using monoclonal antibodies. Eur J Immunol 13:693–700
Edwards BM, Barash SC, Main SH et al (2003) The remarkable flexibility of the human antibody repertoire; isolation of over one thousand different antibodies to a single protein, BLyS. J Mol Biol 334:103–118
Benjamin DC, Berzofsky JA, East IJ et al (1984) The antigenic structure of proteins: a reappraisal. Ann Rev Immunol 2:67–101
Berzofsky JA (1985) Intrinsic and extrinsic factors in protein antigenic structure. Science 229:932–940
Tilley SA, Honnen WJ, Racho ME et al (1992) Synergistic neutralization of HIV-1 by human monoclonal antibodies against the V3 loop and the CD4-binding site of gp120. AIDS Res Hum Retrovir 8:461–467
Laal S, Burda S, Gorny MK et al (1994) Synergistic neutralization of human immunodeficiency virus type 1 by combinations of human monoclonal antibodies. J Virol 68:4001–4008
Zwick MB, Wang M, Poignard P et al (2001) Neutralization synergy of human immunodeficiency virus type 1 primary isolates by cocktails of broadly neutralizing antibodies. J Virol 75:12198–12208
Zhou T, Xu L, Dey B, Hessell AJ et al (2007) Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445:732–737
Zwick MB, Parren PW, Saphire EO et al (2003) Molecular features of the broadly neutralizing immunoglobulin G1 b12 required for recognition of human immunodeficiency virus type 1 gp120. J Virol 77:5863–5876
Loor F (1971) On the existence of heterospecific antibodies in sera from rabbits immunized against tobacco mosaic virus determinants. Immunology 21:557–564
Al Moudallal Z, Briand JP, Van Regenmortel MHV (1982) Monoclonal antibodies as probes of the antigenic structure of tobacco mosaic virus. EMBO J 1:1005–1010
Underwood PA (1985) Theoretical considerations of the ability of monoclonal antibodies to detect antigenic differences between closely related variants, with particular reference to heterospecific reactions. J Immunol Methods 85:295–307
Harper M, Lema F, Boulot G, Poljak RJ (1987) Antigen specificity and cross-reactivity of monoclonal anti lysozyme antibodies. Mol Immunol 24:97–108
Van Regenmortel MHV (1967) Serological studies on naturally occurring strains and chemically induced mutants of tobacco mosaic virus. Virology 31:467–480
Eisen HN (2001) Specificity and degeneracy in antigen recognition: Yin and Yang in the immune system. Ann Rev Immunol 19:1–21
Berzofsky JA, Schechter AN (1981) The concepts of cross-reactivity and specificity in immunology. Mol Immunol 18:751–763
Roberts VA, Getzoff ED, Tainer JA (1993) Structural basis of antigenic cross-reactivity. In: Van Regenmortel MHV (ed) Structure of antigens, vol 2. CRC Press, Boca Raton, pp 31–53
Timmerman P, Puijk WC, Meloen RH (2007) Functional reconstruction and synthetic mimicry of a conformational epitope using CLIPS technology. J Mol Recognit 20:283–299
Pantophlet R, OllmannSaphire E, Poignard P et al (2003) Fine mapping of the interaction of neutralizing and nonneutralizing monoclonal antibodies with the CD4 binding site of human immunodeficiency virus type 1 gp120. J Virol 77:642–658
Parren PW, Burton DR (2001) The antiviral activity of antibodies in vitro and in vivo. Adv Immunol 77:195–262
Van Regenmortel MHV (2006) Immunoinformatics may lead to a reappraisal of the nature of B cell epitopes and of the feasibility of synthetic peptide vaccines. J Mol Recognit 19:183–187
Reading SA, Dimmock NJ (2007) Neutralization of animal virus infectivity by antibody. Arch Virol 152:1047–1059
Burton DR, Pyati J, Koduri R et al (1994) Efficient neutralization of primary isolates of HIV-1 by a recombinant human monoclonal antibody. Science 266:1024–1027
Kwong PD, Wyatt R, Robinson J et al (1998) Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393:648–659
Labrijn AF, Poignard P, Raja A et al (2003) Access of antibody molecules to theconserved coreceptor binding site on glycoprotein gp120 is sterically restricted onprimary human immunodeficiency virus type 1. J Virol 77:10557–10565
Decker JM, Bibollet-Ruche F, Wei X et al (2005) Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J Exp Med 201:1407–1419
Irving MB, Craig L, Menendez A et al (2010) Exploring peptide mimics for the production of antibodies against discontinuous protein epitopes. Mol Immunol 47:1137–1148
Saphire EO, Montero M, Menendez A et al (2007) Structure of a high-affinity “Mimotope” peptide bound to HIV-1-neutralizing antibody b12 explains its inability to elicit gp120 cross-reactive antibodies. J Mol Biol 369:696–709
Sattentau QJ, Moore JP (1995) Human immunodeficiency virus type 1 neutralization is determined by epitope exposure on the gp120 oligomer. J Exp Med 182:185–196
Van Regenmortel MHV (2010) Antigenicity and immunogenicity of viral proteins. In: Mahy BWJ, Van Regenmortel MHV (eds) Desk encyclopedia of general virology. Academic Press, Elsevier, Oxford, pp 343–349
Berek C (1992) Maturation of the immune response. In: Van Regenmortel MHV (ed) Structure of antigens, vol 1. CRC Press, Boca Raton, pp 149–157
Harwood NE, Batista FD (2010) Early events in B cell activation. Ann Rev Immunol 28:185–210
Rini JM, Schulze-Gahmen U, Wilson IA (1992) Structural evidence for induced fit as a mechanism for antibody-antigen recognition. Science 255:959–965
Wilson IA, Stanfield RL (1994) Antigen-antibody interactions: new structures and new conformational changes. Curr Opin Struct Biol 4:857–867
Berger C, Weber-Bornhauser S, Eggenberger J, Hanes J, Plückthun A, Bosshard HR (1999) Antigen recognition by conformational selection. FEBS Lett 450:149–153
Bosshard HR (2001) Molecular recognition by induced fit: how fit is the concept? News Physiol Sci 16:171–173
Goh CS, Milburn D, Gerstein M (2004) Conformational changes associated with protein-protein interactions. Curr Opin Struct Biol 14:104–109
Pantophlet R, Burton DR (2003) Immunofocusing: antigen engineering to promote the induction of HIV-neutralizing antibodies. Trends Mol Med 9:468–473
Wu X, Yang ZY, Li Y, Hogerkorp CM et al (2010) Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861
Joyce JG, Hurni WM, Bogusky MJ et al (2002) Enhancement of alpha-helicity in the HIV-1 inhibitory peptide DP178 leads to an increased affinity for human monoclonal antibody 2F5 but does not elicit neutralizing responses in vitro. J Biol Chem 277:45811–45820
Ho J, Uger RA, Zwick MB et al (2005) Conformational constraints imposed on a pan-neutralizing HIV-1 antibody epitope result in increased antigenicity but not neutralizing response. Vaccine 23:1559–1573
Hans D, Young PR, Fairlie DP (2006) Current status of short synthetic peptides as vaccines. Med Chem 2:627–646
Thornton JM, Sibanda BL (1983) Amino and carboxy-terminal regions in globular proteins. J Mol Biol 167:443–460
Westhof E, Altschuh D, Moras D et al (1984) Correlation between segmental mobility and the location of antigenic determinants in proteins. Nature 311:123–126
Tainer JA, Getzoff ED, Paterson Y et al (1985) The atomic mobility component of protein antigenicity. Ann Rev Immunol 3:501–535
Pellequer J-L, Westhof E, Van Regenmortel MHV (1994) Epitope predictions from the primary structure of proteins. In: Wisdow GB (ed) Peptide antigens: a practical approach. JRL, Oxford, pp 7–25
Leinikki P, Lehtinen M, Hyöty H et al (1993) Synthetic peptides as diagnostic tools in virology. Adv Vir Res 42:149–186
Walter G (1986) Production and use of antibodies against synthetic peptides. J Immunol Methods 88:149–161
Muller S (1999) Use of antipeptide antibodies in molecular and cellular biology. In: Van Regenmortel MHV, Muller S (eds) Synthetic peptide as antigens. Elsevier Amsterdam, The Netherlands, pp 215–235
Blythe MJ, Flower DR (2005) Benchmarking B cell epitope prediction: Under performance of existing methods. Protein Sci 14:246–248
Caioli SE (2010) Benchmarking B-cell epitope prediction for the design of peptide-based vaccines: problems and prospects. J Biomed Biotechnol, vol 2010, article ID 910524, doi:10.1155/2010/910524
Langeveld JP, Casal JI, Osterhaus AD et al (1994) First peptide vaccine providing protection against viral infection in the target animal: studies of canine parvovirus in dogs. J Virol 68:4506–4513
Bittle JL, Hougten RA, Alexander H et al (1982) Protection against foot-and-mouth disease by immunization with a chemically synthesized peptide predicted from the viral nucleotide sequence. Nature 298:30–33
Rowlands DJ, Clarke BE, Carroll AR et al (1983) Chemical basis of antigenic variation in foot-and-mouth disease virus. Nature 306:694–697
Parry NR, Barnett PV, Ouldridge EJ et al (1989) Neutralizing epitopes of type O foot-and-mouth disease virus. II. Mapping three conformational sites with synthetic peptide reagents. J Gen Virol 70:1493–1503
Uversky VN, Oldfield CJ, Dunker AK (2005) Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit 18:343–384
Spear GR, Takefman DM, Sharpe S et al (1994) Antibodies to the HIV-1 V3 loop in serum from infected persons contribute a major proportion of immune effect or functions including complement activation, antibody binding, and neutralization. Virology 204:609–615
Jiang X, Burke V, Totrov M et al (2010) Conserved structural elements in the V3 crown of HIV-1 gp120. Nat Struct Mol Biol 17:955–961
VanCott TC, Polonis VR, Loomis LD et al (1995) Differential role of V3-specific antibodies in neutralization assays involving primary and laboratory-adapted isolates of HIV type 1. AIDS Res Hum Retrovir 11:1379–1391
Kayman SC, Wu Z, Revesz K et al (1994) Presentation of native epitopes in the V1/V2 and V3 regions of human immunodeficiency virus type 1gp120 by fusion glycoproteins containing isolated gp120 domains. J Virol 68:400–410
Gorny MK, Williams C, Volsky B et al (2006) Cross-clade neutralizing activity of human anti-V3 monoclonal antibodies derived from the cells of individuals infected with non-B clades of human immunodeficiency virus type. J Virol 80:6865–6872
Stanfield R, Cabezas E, Satterthwait A et al (1999) Dual conformations for the HIV-1 gp120 V3 loop in complexes with different neutralizing fabs. Structure 7:131–142
Rosen O, Chill J, Sharon M et al (2005) Induced fit in HIV-neutralizing antibody complexes: evidence for alternative conformations of the gp120 V3 loop and the molecular basis for broad neutralization. Biochemistry 44:7250–7258
Mester B, Manor R, Mor A et al (2009) HIV-1 peptide vaccine candidates: selecting constrained V3 peptides with highest affinity to antibody 447–52D. Biochemistry 48:7867–7877
Briand JP, Benkirane N, Guichard G et al (1997) A retro-inverso peptide corresponding to the GH loop of foot-and-mouth disease virus elicits high levels of long-lasting protective neutralizing antibodies. Proc Natl Acad Sci USA 94:12545–12550
Carver JA, Esposito G, Viglino P et al (1997) Structural comparison between retro-inverso and parent peptides: molecular basis for the biological activity of a retro-inverso analogue of the immunodominant fragment of VP1 coat protein from foot-and-mouth disease virus. Biopolymers 41:569–589
Nargi F, Kramer E, Mezencio J et al (1999) Protection of swine from foot-and-mouth disease with one dose of an all-D retro peptide. Vaccine 17:2888–2893
Hilleman MR (1992) Impediments, imponderables and alternatives in the attempt to develop an effective vaccine against AIDS. Vaccine 10:1053–1058
Kusters I, Almond JW (2010) Vaccine strategies. In: Mahy BWJ, Van Regenmortel MHV (eds) Desk encyclopedia of general virology. Academic Press, Elsevier, Oxford, pp 381–389
Virgin HW, Walker BD (2010) Immunology and the elusive AIDS vaccine. Nature 464:224–231
Scheid JF, Mouquet H, Feldhahn N et al (2009) Broad diversity of neutralizing antibodies isolated from memory B cells in HIV-infected individuals. Nature 458:636–640
Walker LM, Phogat SK, Chan-Hui P et al (2009) Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289
Corti D, Langedijk JP, Hinz A et al (2010) Analysis of memory B cell responses and isolation of novel monoclonal antibodies with neutralizing breadth from HIV-1-infected individuals. PLoS one 5:e8805
Mascola JR, Montefiori DC (2010) The role of antibodies in HIV vaccines. Annu Rev Immunol 28:413–444
Nara PL, Garrity R (1998) Deceptive imprinting: a cosmopolitan strategy for complicating vaccination. Vaccine 16:1780–1787
Tobin GJ, Trujillo JD, Bushnell RV et al (2008) Deceptive imprinting and immune refocusing in vaccine design. Vaccine 26:6189–6199
Lu S, Wyatt R, Richmond JF et al (1998) Immunogenicity of DNA vaccines expressing human immunodeficiency virus type 1 envelope glycoprotein with and without deletions in the V1/2 and V3 regions. AIDS Res Hum Retrovir 14:151–155
Kim YB, Han DP, Cao C, Cho MW (2003) Immunogenicity and ability of variable loop-deleted human immunodeficiency virus type 1 envelope glycoproteins to elicit neutralizing antibodies. Virology 305:124–137
Gaschen B, Taylor J, Yusim K et al (2002) Diversity considerations in HIV-1 vaccine selection. Science 296:2354–2360
McBurney SP, Ross TM (2009) Human immunodeficiency virus-like particles with consensus envelopes elicited broader cell-mediated peripheral and mucosal immune responses than polyvalent and monovalent Env vaccines. Vaccine 27:4337–4349
Gras-Masse H, Georges B, Estaquier J et al (1999) Convergent peptide libraries, or mixotopes, to elicit or to identify specific immune responses. Curr Opin Immunol 11:223–228
Korber B, Gnanakaran S (2009) The implications of patterns in HIV diversity for neutralizing antibody induction and susceptibility. Curr Opin HIV AIDS 4:408–417
Pantophlet R, Aguilar-Sino RO, Wrin T et al (2007) Analysis of the neutralization breadth of the anti-V3 antibody F425–B4e8 and re-assessment of its epitope fine specificity by scanning mutagenesis. Virology 364:441–453
Bramwell VW, Perrie Y (2005) The rational design of vaccines. Drug Disc Today 10:1527–1534
D’Argenio DA, Wilson CB (2010) A decade of vaccines: Integrating immunology and vaccinology for rational vaccine design. Immunity 33:437–440
McElrath MJ, Haynes BF (2010) Induction of immunity to human immunodeficiency virus type-1 by vaccination. Immunity 33:542–554
Sette A, Rappuoli R (2010) Reverse vaccinology: vaccines in the era of genomics. Immunity 33:530–541
Obeid OE, Partidos CD, Howard CR, Steward MW (1995) Protection against morbillivirus-induced encephalitis by immunization with a rationally designed synthetic peptide vaccine containing B- and T-cell epitopes from the fusion protein of measles virus. J Virol 69:1420–1428
Bunge M (1993) Philosophical dictionary. Prometheus Books, Amherst, New York
Van Regenmortel MHV (2007) The rational design of biological complexity: a deceptive metaphor. Proteomics 7:965–975
Van Regenmortel MHV (2002) Reductionism and the search for structure-function relationships in antibody molecules. J Mol Recognit 15:240–247
Zhou T, Georgiev I, Wu X et al (2010) Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329:811–817
Binley JM, Lybarger EA, Crooks ET et al (2008) Profiling the specificity of neutralizing antibodies in a large panel of plasmas from patients chronically infected with human immunodeficiency virus type 1 subtypes B and C. J Virol 82:11651–11658
Gray ES, Taylor N, Wycuff D et al (2009) Antibody specificities associated with neutralization breadth in plasma from human immunodeficiency virus type 1 subtype C-infected blood donors. J Virol 83:8925–8937
Burton DR, Weiss RA (2010) A boost for HIV vaccine design. Science 329:770–773
Ebensen T, Guzmàn CA (2008) Immune modulators with defined molecular targets: cornerstone to optimize rational vaccine design. Hum Vaccin 4:13–22
Tobin MB, Gustafsson C, Huisman GW (2000) Directed evolution: the ‘rational’ basis for ‘irrational’ design. Curr Opin Struct Biol 10:421–427
Villén J, Rodríguez-Mias RA, Núñez JI et al (2006) Rational dissection of binding surfaces for mimicking of discontinuous antigenic sites. Chem Biol 13:815–823
Poropatich K, Sullivan DJ Jr (2011) Human immunodeficiency virus type 1 long-term non-progressors: the viral, genetic and immunological basis for disease non-progression. J Gen Virol 92:247–268
Okulicz JF, Marconi VC, Landrum ML et al (2009) Clinical outcomes of elite controllers, viremic controllers, and long-term nonprogressors in the US Department of Defense HIV natural history study. J Infect Dis 200:1714–1723
Simek MD, Rida W, Priddy FH et al (2009) Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol 83:7337–7348
Mascola JR (1999) Herpes simplex virus vaccines–why don’t antibodies protect? J Am Med Assoc 281:379–380
Pulendran B, Li S, Nakaya HI (2010) System vaccinology. Immunity 33:516–529
García F, Climent N, Assoumou L et al (2011) A therapeutic dendritic cell-based vaccine for HIV-1 infection. J Infect Dis 203:473–478
Li JZ, Brumme ZL, Brumme CJ et al (2011) Factors associated with viral rebound in HIV-1-infected individuals enrolled in a therapeutic HIV-1 gag vaccine trial. J Infect Dis 203:976–983
Corey L, Nabel GJ, Dieffenbach C et al (2011) HIV-1 vaccines and adaptive trial designs. Sci Trans Med 3: 79ps13
Burton DR, Desrosiers RC, Doms RW et al (2004) A sound rationale needed for phase III HIV-1 vaccine trials. Science 303:316
McNeil JG, Johnston MI, Birx DL, Tramont EC (2004) HIV vaccine trial justified. Science 303:961
Belshe R, Franchini G, Girard MP et al (2004) Support for the RV144 HIV vaccine trial. Science 305:177–180
Jefferys R, Harrington M (2004) Outstanding questions on HIV vaccine trial. Science 305:180
Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S et al (2009) Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. New Engl J Med 361:2209–2220
Letvin NL (2009) Moving forward in HIV vaccine development. Science 326:1196–1198
Acknowledgments
The author is grateful to Pernille Haste-Anderson of the Danish Technically University, Denmark for preparing Figure 1 (A, B) and to Florence Diemer for efficient secretarial assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Van Regenmortel, M.H.V. Requirements for empirical immunogenicity trials, rather than structure-based design, for developing an effective HIV vaccine. Arch Virol 157, 1–20 (2012). https://doi.org/10.1007/s00705-011-1145-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00705-011-1145-2