
Abstract
Brachytelephalangic chondrodysplasia punctata (CDPX1, OMIM: #302950) is a rare congenital skeletal dysplasia caused by arylsulfatase E deficiency (OMIM: #300180). Although the symptoms are usually mild, severe spinal cord compression by dysplastic vertebras may develop. We report four new cases with severe cervical spinal canal narrowing documented by radiography, magnetic resonance imaging (MRI), and autopsy. In all, nine cases of CDPX1 with severe cervical spinal cord compression have now been described. Because these cases account for a large proportion of all reported CDPX1 cases, we believe that an antenatal suspicion of CDPX1 should lead to genetic counseling and to investigations for spinal cord compression. After birth, this complication must be routinely anticipated, and we suggest spinal MRI in all CDPX1 infants. Unless spinal cord compression is confidently ruled out, we recommend that these newborns receive the same care as trauma patients suspected of craniocervical junction disruption.
Similar content being viewed by others

Introduction
Brachytelephalangic chondrodysplasia punctata (CDPX1, OMIM #302950), which was first described by Curry et al. in 1984 [3], is a member of a group of diseases characterized by dysplastic cartilage [1, 9, 12]. CDPX1 is inherited as an X-linked recessive trait and is due to a mutation in the ARSE gene (ARSE, OMIM: #300180) encoding the enzyme arylsulfatase E and located at Xp22.32. The prevalence of this rare condition is only 1 per 500,000 newborns [2, 8, 10]. The clinical presentation includes hypoplasia of the nose and of the distal phalanges (brachytelephalangia). Radiographs show multiple, premature, punctate calcifications of the epiphyses. Although the manifestations are usually mild, a dreaded complication is severe cervical myelopathy caused by spinal dislocation or spinal canal stenosis [4, 6, 7].
We report four new cases of CDPX1 with myelopathy, including three diagnosed antenatally. Severe cervical compression was confirmed at autopsy in three cases.
Case reports
Case 1
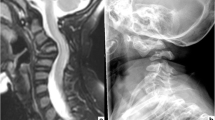
This boy was the third child of unrelated African parents who had an unremarkable medical history. The pregnancy was free of drug exposure. Nasal hypoplasia was noted by antenatal ultrasonography (US). At 39 weeks of gestation (WOG), the infant was delivered vaginally in meconium-stained amniotic fluid and developed respiratory distress, requiring admission to the neonatal intensive care unit (NICU). The physical examination showed nasal hypoplasia with a flat nasal bridge and barely patent nostrils (Fig. 1a). On the second postnatal day (D2), the neurological examination revealed diffuse pyramidal syndrome and absent bulbopontine reflexes. Radiographs of the skeleton showed diffuse punctate calcifications of the vertebras, brachytelephalangia of both hands (Fig. 1b), and incomplete ossification of several cervical vertebras, with suspected craniovertebral and atlantoaxial dislocations (Fig. 1c). There was no rhizomelic shortening of the tubular bones. Magnetic resonance imaging (MRI) confirmed the vertebral dislocations and showed spinal cord compression at the craniovertebral junction (Fig. 1d), without supratentorial abnormalities. Findings were normal from cardiac, transfontanellar, lower spinal cord, and abdominal US, as well as from an ophthalmologic evaluation and examination of the skin. The karyotype was 46, XY. CDPX1 was considered. Continuous cervical traction was started, but failed to improve the neurological symptoms. Surgery was deemed inappropriate, given the severe manifestations and minimal expected benefits. The autonomic nervous system abnormalities worsened gradually, and the patient died on day 15. The parents declined autopsy.

a Front view of case 1: severely depressed nasal bridge and hypoplastic alar cartilages. b Frontal view of the right hand of case 1. Hypoplastic distal phalanges of the 1st, 3rd, and 4th fingers. c Lateral projection of the cervical spine in case 1. Cervical kyphosis with stippling of the cervical vertebras; the bodies of C3, C4, and C5 are not visible. Craniovertebral and atlantoaxial dislocations are seen. d Magnetic resonance imaging (MRI): T2-weighted sequence, midline sagittal slice of the craniovertebral junction, and cervical spine of the first patient. Note the cervical kyphosis with marked narrowing of the foramen magnum and kinking of the bulbomedullary junction. The perimedullary space is not visible
Case 2
This boy was the first child of unrelated Caucasian parents who had no significant medical history. The pregnancy was unremarkable until 32 WOG, when a sonogram showed nasal hypoplasia, stippling of multiple epiphyses, and polyhydramnios. The karyotype was 46, XY. After vaginal delivery at 34 WOG, the infant was admitted to the NICU for mild respiratory distress. He had nasal hypoplasia with a flat nasal bridge and barely patent nostrils, brachytelephalangia, and thoracic scoliosis. The neurological examination showed tetraparesis. Skeletal radiographs confirmed the presence of multiple premature punctate calcifications of the epiphyses, without shortening of tubular bones. CDPX1 was considered. The neurological deficit worsened gradually and the tetraplegia was complete by 2 months of age. Findings were normal from transfontanellar US, cerebral MRI, and electroencephalograph (EEG). MRI of the spine showed severe stenosis of the cervicooccipital junction, with spinal cord compression and hypoplasia. At 3 months of age, the patient died after life-support was withdrawn during another episode of respiratory distress. The autopsy confirmed cervical spinal cord compression due to spinal canal stenosis. Molecular analysis of the ARSE gene performed as described by Franco et al. [5] showed a point mutation (G355S) in exon 7, confirming the diagnosis of CDPX1.
Cases 3 and 4
Two pregnancies in the same woman were terminated because of suspected CDPX1 with spinal cord compression due to cervical spinal canal stenosis, which was confirmed at autopsy. The parents were unrelated and had no history of significant health problems. CDPX1 was confirmed when the ARSE gene mutation analysis of samples from both fetuses showed a point mutation in exon 10, resulting in a T481M amino acid substitution.
In case 3, the mother was 24 years old. US performed at 22 WOG showed nasal hypoplasia, clubbed feet, punctate calcifications of the femoral and humeral epiphyses, and suspected spinal canal stenosis. There was no rhizomelic shortening of the tubular bones. Fetal biometry was normal. These abnormalities were confirmed at 27 WOG by intra-uterine radiographs, showing numerous vertebral calcifications. The karyotype was 46, XY. CDPX1 with spinal canal stenosis and severe spinal cord compression was strongly suspected. After genetic counselling, the pregnancy was terminated at 27 WOG. Ex-utero radiographs of the entire spine (Fig. 2a) and transverse radiographs of the cervical vertebras (Fig. 2b) showed marked spinal canal stenosis and diffuse stippling of the vertebras and proximal femurs.

a Frontal view of the spine after the termination of pregnancy in case 3, at 27 weeks. Diffuse stippling of the vertebras and proximal femurs. b Transverse radiographs of C7 vertebra in case 3. c Transverse radiographs of C7 in an age-matched control fetus
This woman had a second pregnancy at 28 years of age. At 21 WOG, US showed a male fetus with the same facial and spinal abnormalities as the first fetus. These findings were confirmed at 25 WOG, when US also showed stenosis of the distal cervical spinal canal and brachytelephalangia. The karyotype was 46, XY. After genetic counselling, the pregnancy was terminated at 26 WOG.
Discussion
Although the CDPX1 phenotype is variable [2], our four cases had typical clinical and radiological features, with nasal hypoplasia, brachytelephalangia, and multiple punctate calcifications of the epiphyses and vertebras. The absence of rhizomelic shortening of tubular bones, bilateral cataract, cutaneous abnormalities, or growth asymmetry ruled out other forms of chondrodysplasia [12]. Moreover, the identification of point mutations of the ARSE gene in three cases confirmed the diagnosis of CDPX1. The T481M mutation observed in cases 3 and 4 has been previously reported by Brunetti-Pierri et al. [2]. Expression in cos cells showed that the mutation led to arylsulfatase E deficiency, indicating that it probably causes the disease. The G355S mutation found in case 2 has not been described earlier. No functional analysis of this mutation is available. However, the glycine residue at position 355 is highly conserved in human sulfatases, as described by Sardiello et al. [11]. This strongly supports a causal role of the mutation in the disease.
Including our four new cases, nine cases of CDPX1 with severe cervical spinal cord compression have now been reported. Given the small total number of reported CDPX1 cases, these numbers indicate that spinal cord compression may be a common manifestation [4, 6, 7]. Therefore, we recommend genetic counselling when CDPX1 is suspected antenatally.
Spinal canal stenosis may be suspected by antenatal US, as shown by our cases 3 and 4. Antenatal MRI of the spinal cord may be more sensitive for detecting spinal cord compression, but no data are available on this point. The clinical diagnosis seems difficult in the first postnatal days. In our case 2 and in two other reported cases, spinal cord compression was diagnosed at 2 months of age [4, 7]. Ventilator weaning difficulties in patients with CDPX1 should alert neonatologists to the possibility of spinal cord compression. Alternatively, they may indicate upper airway malformations with aberrant localization of calcifications, as previously reported [13]. However, central hypoventilation caused by cervical spinal cord compression must be searched for.
Conclusion
Spinal cord compression should be sought routinely in patients with brachytelephalangic chondrodysplasia punctata (CDPX1). To this end, we suggest routine magnetic resonance imaging (MRI) of the spine. Until CDPX1 is confidently ruled out, we recommend the same care as in trauma patients with suspected craniocervical junction disruption.
Abbreviations
- CDPX1:
-
brachytelephalangic chondrodysplasia punctata
- MRI:
-
magnetic resonance imaging
- ARSE:
-
arylsulfatase E
- US:
-
ultrasound
- WOG:
-
weeks of gestation
- NICU:
-
Neonatal intensive care unit
References
Bennett CP, Berry AC, Maxwell DJ, Seller MJ (1992) Chondrodysplasia punctata: another possible X-linked recessive case. Am J Med Genet 44(6):795–799
Brunetti-Pierri N, Andreucci MV, Tuzzi R, Vega GR, Gray G, McKeown C, Ballabio A, Andria G, Meroni G, Parenti G (2003) X-linked recessive chondrodysplasia punctata: spectrum of arylsulfatase E gene mutations and expanded clinical variability. Am J Med Genet A 117(2):164–168
Curry CJ, Magenis RE, Brown M, Lanman JT Jr, Tsai J, O’Lague P, Goodfellow P, Mohandas T, Bergner EA, Shapiro LJ (1984) Inherited chondrodysplasia punctata due to a deletion of the terminal short arm of an X chromosome. N Engl J Med 311(16):1010–1015
Eash DD, Weaver DD, Brunetti-Pierri N (2003) Cervical spine stenosis and possible vitamin K deficiency embryopathy in an unusual case of chondrodysplasia punctata and an updated classification system. Am J Med Genet A 122(1):70–75
Franco B, Meroni G, Parenti G, Levilliers J, Bernard L, Gebbia M, Cox L, Maroteaux P, Sheffield L, Rappold GA (1995) A cluster of sulfatase genes on Xp22.3: mutations in chondrodysplasia punctata (CDPX) and implications for warfarin embryopathy. Cell 81(1):15–25
Goldstein J, Arbelaez AM, Bismar T, Grange DK, Martin RA (2001) Cervical stenosis and cord compression in brachytelephalangic chondrodysplasia punctata: A previously unreported severe complication. XXII David W. Smith Workshop on Malformations and Morphogenesis, Lake Arrowhead, California, September 2001, p 109
Herman TE, Lee BC, McAlister WH (2002) Brachytelephalangic chondrodysplasia punctata with marked cervical stenosis and cord compression: report of two cases. Pediatr Radiol 32(6):452–456
Malou E, Gekas J, Troucelier-Lucas V, Mornet E, Razafimanantsoa L, Cuvelier B, Mathieu M, Thepot F (2002) X-linked recessive chondrodysplasia punctata. Cytogenetic study and role of molecular biology. Arch Pediatr 8(2):176–180
Maroteaux P (1989) Brachytelephalangic chondrodysplasia punctata: a possible X-linked recessive form. Hum Genet 82(2):167–170
Muroya K, Ogata T, Rappold G, Klink A, Nakahori Y, Fukushima Y, Aizu K, Matsuo N (1995) Refinement of the locus for X-linked recessive chondrodysplasia punctata. Hum Genet 95(5):577–580
Sardiello M, Annunziata I, Roma G, Ballabio A (2005) Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum Mol Genet 14(21):3203–3217
Wessels MW, Den Hollander NJ, De Krijger RR, Nikkels PG, Brandenburg H, Hennekam R, Willems PJ (2003) Fetus with an unusual form of nonrhizomelic chondrodysplasia punctata: case report and review. Am J Med Genet A 120(1):97–104
Wolpoe ME, Braverman N, Lin SY (2004) Severe tracheobronchial stenosis in the X-linked recessive form of chondrodysplasia punctata. Arch Otolaryngol Head Neck Surg 130(12):1423–1426
Author information
Authors and Affiliations
Corresponding author
Additional information
No conflicts of interest occurred for this work.
Rights and permissions
About this article
Cite this article
Garnier, A., Dauger, S., Eurin, D. et al. Brachytelephalangic chondrodysplasia punctata with severe spinal cord compression: report of four new cases. Eur J Pediatr 166, 327–331 (2007). https://doi.org/10.1007/s00431-006-0239-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00431-006-0239-4