
Abstract
TRPC5 is a member of the canonical transient receptor potential (TRPC) family of proteins that forms cationic channels either through homomultimeric assembly or heteromultimeric coordination with other TRPC proteins. It is expressed in a variety of cells including central neurones and endothelial cells and has susceptibility to stimulation by multiple factors. Here we investigated if TRPC5 is sensitive to nitric oxide. Mouse TRPC5 or human TRPC5 was over-expressed in HEK293 cells, and TRPC5 activity was determined by measuring the cytosolic Ca2+ concentration with an indicator dye or by recording membrane current under voltage clamp. TRPC5 activity could be evoked by carbachol acting at muscarinic receptors, lanthanum, or a reducing agent. However, S-nitroso-N-acetylpenicillamine (SNAP) and diethylamine NONOate (DEA-NONOate) failed to stimulate or inhibit TRPC5 at concentrations that generated nitric oxide, caused vasorelaxation, or suppressed activity of TRPC6 via protein kinase G. At high concentrations, SNAP (but not DEA-NONOate) occasionally stimulated TRPC5 but the effect was confounded by background TRPC5-independent Ca2+ signals. Endogenous Ca2+-entry in bovine aortic endothelial cells (BAECs) was suppressed by SNAP; TRPC5 blocking antibody or dominant-negative mutant TRPC5 suppressed this Ca2+ entry and occluded the effect of SNAP. The data suggest that nitric oxide is not a direct modulator of homomeric TRPC5 channels but may inhibit endogenous BAEC channels that contain TRPC5.
Similar content being viewed by others


Introduction
Nitric oxide (NO) is a short-lived, reactive molecule that serves as an important cellular signal [15]. NO signaling is involved in endothelial-dependent vascular relaxation, cell motility, immune defense, and neurite outgrowth [2, 4, 7]. While many NO-elicited responses are mediated by cGMP, which is produced from the activity of NO-stimulated guanylate cyclase [15], NO can also act independently of cGMP [6]. A well-documented NO action independent of cGMP is S-nitrosylation. In S-nitrosylation, a nitric oxide moiety is covalently attached to cysteine residues of a protein, resulting in alteration in protein function. S-nitrosylation serves as a type of posttranslational modification analogous to phosphorylation and acetylation. While some substrate proteins are constitutively S-nitrosylated [8], other proteins can be reversibly S-nitrosylated dependent upon the concentration of NO and the accessibility of the NO radicals to the cysteine residues [6]. A wide spectrum of proteins including metabolic, structural, and signaling proteins can be S-nitrosylated [6, 8]. S-nitrosylation is reported to modulate the activity of a number of ion channels including Ca2+-activated K+ channels, ryanodine receptors, hyperpolarization-activated cation channels, cyclic nucleotide-gate channels, and N-methyl-D-aspartate receptors [6, 8, 16].
TRPC5 is a Ca2+-permeable cationic channel that modulates properties of mammalian cells including neurones and endothelial cells [1]. It can form channels on its own or assembled with other related proteins such as TRPC1 and TRPC4. Each channel is thought to require four TRPC proteins to assemble around a central ion pore. The channels show constitutive activity and voltage dependence but they are also stimulated by a range of factors including agonists at G protein-coupled receptors, lysophospholipids, redox factors, and acidification. In addition, it has been suggested that the channels are stimulated by NO through a mechanism that requires direct S-nitrosylation of TRPC5 at two cysteine residues near the ion pore [24]. The effect was suggested to be functionally important in endothelial cells, which have a crucial role in generating NO. However, the conclusions are difficult to reconcile with data suggesting that the same cysteine residues form a disulphide bridge [23] and that NO has commonly been found to be an inhibitor of Ca2+ entry in endothelial cells [5, 11, 18, 21, but see 13]. Here, we show data generated independently by our two laboratories that lead us to conclude that NO is not a direct modulator of TRPC5.
Materials and methods
Cell culture and cDNA expression
For single-cell Ca2+ imaging studies, mouse TRPC5 (mTRPC5) in pcDNA3 or human TRPC3 (hTRPC3) in pcDNA6 were stably transfected into HEK293 cells. For experiments with protein kinase G (PKG) inhibition of TRPC6, PKG-1α in pcDNA6 and mTRPC6 in pcDNA3 were stably cotransfected into HEK293 cells. mTRPC5 and mouse TRPC6 clones were gifts from L. Birnbaumer (NIH). Transfection and cell culture of HEK293 were done as described elsewhere [12]. Expression of TRPC5 was validated by western blotting (Suppl. Fig. 1). For the multiwell fluorescence plate reader system, HEK293 cells stably expressing tetracycline-inducible human TRPC5 (hTRPC5) was established and cultured as described previously [25]. The expression of hTRPC5 was induced by 1 µg/ml tetracycline (Tet+) for 24–48 h before experiments. Noninduced cells without addition of tetracycline (Tet−) were used as control. The primary cultured bovine aortic endothelial cells (BAECs) were isolated from bovine aorta obtained from the local abattoir and using methodology described previously [3]. Briefly, aortic segments were cut open longitudinally. The intima layer was peeled off and then digested with 0.1% collagenase in phosphate-buffered saline (PBS; in mM: 140 NaCl, 3 KCl, 25 Tris, pH 7.4) for 15 min at 37°C under vigorous shaking. Dissociated cells were centrifuged, resuspended, and then grown in a culture medium RPMI-1640 supplemented with 10% fetal bovine serum. Cells of passage 3–12 were used for experiments. In some experiments, BAECs were transfected with pEGFP-N1 or dominant-negative TRPC5 construct (DN-T5) by electroporation (Nucleofector, Lonza Cologne). Transfected cells were seeded on coverslips and cultured for 2 days before experiments. DN-T5 was gift from D. Clapham (Harvard University). All cells were cultured in 95% air and 5% CO2.
Intracellular Ca2+-measurement
For single-cell Ca2+ imaging studies, intracellular Ca2+ ([Ca2+]i) was imaged using the InCyt Basic Im2 live cell system (Intracellular Imaging). For multiwell intracellular Ca2+ measurement studies, intracellular Ca2+ was measured using a fluorescence plate reader system (FlexStation, Molecular Devices). In single-cell Ca2+ imaging experiments, cells seeded on coverslip were loaded with 5 µM fura-2 AM in culture medium for 30 min and then mounted in a recording chamber containing bath solution. Stock solutions of the chemicals were kept on ice during the experiment. At approximately 10 s before each chemical addition, one tenth of the bath solution was pipetted to dilute the chemical from its stock, before readdition of the solution to the bath. Normal physiological saline solution contained, in mM: 140 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 10 glucose, 10 Hepes (pH titrated to 7.4 with NaOH). Hepes-buffered saline (HBS) contained in mM: 107 NaCl, 6 KCl, 1.2 MgSO4, 2 CaCl2, 11.5 glucose, 20 Hepes (pH titrated to 7.4 with NaOH). HBS-EGTA contained in mM: 107 NaCl, 6 KCl, 1.2 MgSO4, 0.5 EGTA, 11.5 glucose, 20 Hepes (pH titrated to 7.4 with NaOH). For FlexStation experiments, cells were plated in 96-well biocoat plates (Corning) to 80–90% confluence 24–48 h before experiments. Prior to recording, cells were incubated for 1 h at 37°C in standard bath solution (SBS) containing 2 µM fura-2 AM and (in mM) 130 NaCl, 5 KCl, 8 glucose, 10 Hepes, 1.2 MgCl2, and 1.5 CaCl2 (pH titrated to 7.4 with NaOH). Subsequently, the cells were washed with fresh SBS for 0.5 h at room temperature. Fura-2 was excited by light at 340 and 380 nm and emission was filtered at 510 nm. Experiments were at room temperature (∼23°C). Most experiments were performed using fura-2 as the fluorescent dye, where the [Ca2+]i response was expressed as the ratio of the fluorescence signal from excitation at 340 nm to that at 380 nm (F340/380). Some experiments were done using fluo-3 as the fluorescent dye, where the [Ca2+]i response was expressed as real-time fluo-3 fluorescence relative to its initial value before application of chemicals (Ft/F0). In FlexStation experiments, NO donors were diluted in bath solution to five times the final concentration immediately before each experiment and applied to cells at five times dilution by the FlexStation within 5 (column 1) and 30 min (column 2); no difference between data from columns 1 and 2 was observed.
Electrophysiology
To study hTRPC5, whole-cell patch-clamp recordings were performed as described previously [23]. Patch pipette solution contained (in mM): 115 CsCl, 10 EGTA, 2 MgCl2, 5 Na2ATP, 0.1 NaGTP, 10 Hepes, 5.7 CaCl2 (osmolarity was adjusted to ∼290 mOsm with mannitol and pH was titrated to 7.2 using CsOH). The extracellular (bath) solution was SBS. A salt–agar bridge was used to connect the Ag–AgCl ground wire to the bath. Signals were amplified with an Axopatch 200B patch-clamp amplifier and controlled with Signal software 3.05 (CED). A 1-s ramp protocol from −100 mV to +100 mV was applied at a frequency of 0.1 Hz from a holding potential of −60 mV. Current signals were analogue filtered (Bessel) at 1 kHz and digitally sampled at 3 kHz. NO donors were prepared freshly for each experiment within 1–2 min before application.
Isometric tension measurement
Segments of mouse aorta ∼2 mm in length were dissected from ∼5-week-old male C57BL/6 mice and the endothelial layer was rubbed off. The segments were mounted in a wire myograph (610 M, Danish Myograph Technology) under a normalized tension as previously described [3]. The aortic segments were preconstricted with 10 µM phenylephrine to achieve sustained contractions. Approximately 30 min before phenylephrine-induced precontraction, dimethyl sulfoxide (DMSO)-diluted NO donors were prepared from stocks into the following concentrations (in mM): 0.1, 0.3, 1, and 300 for SNAP; 0.01, 0.1, 1, 10, and 100 for diethylamine NONOate (DEA-NONOate). The diluted NO donors were kept on ice and used for a single set of experimental recordings. The NO donors were added in a cumulative fashion to the bath solution, which was Krebs solution containing, in mM: 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25.2 NaHCO3, and 11.1 glucose, gassed with 95% O2–5% CO2. Experiments were performed at 37°C. Data were acquired and analyzed using PowerLab and LabChart (AD Instruments).
Measurement of NO release using multiwell imaging system
HEK293 cells cultured in 96-well plates were loaded with 10 µM DAF-FM acetate (Invitrogen, UK) by incubating at room temperature for 45 min. Cells were washed for 20 min and fluorescence emission at 520 nm was measured with excitation at 495 nm by the multiwell fluorescence plate reader system (FlexStation, Molecular Devices). Fluorescence values were divided by 1,000 and indicated as F*.
Immunoblotting
Whole-cell lysates were extracted with detergent extraction buffer, which contained 1% (v/v) Nonidet P-40, 150 mM NaCl, 20 mM Tris–HCl, pH 8.0, or NuPAGE LDS buffer (Invitrogen) with addition of protease inhibitor cocktail tablets. Equal quantities of extracted proteins were applied to each lane and separated on SDS/PAGE gel. The proteins were then blotted onto a PVDF/nitrocellulose membrane. The membrane was incubated at 4°C overnight with anti-TRPC5 or anti-TRPC6 antibody (dilution 1:200 for Alomone Lab anti-TRPC5 and anti-TRPC6, or dilution 1:500 for custom made T5C3 to peptide CKLLDSSEDVFETWGE) in PBS solution (Invitrogen) containing 0.1% Tween 20 and 5% nonfat dry milk. The membrane was washed in PBS + Tween 20 (0.1%) and incubated with horseradish-conjugated secondary antibodies for 1 h. Immunodetection was accomplished by ECL detection system and X-ray film exposure. Immunoblots with anti-β-tubulin antibody or anti-β-actin (Santa Cruz Biotechnology) were used to confirm that an equal amount of proteins was loaded onto each lane.
Immunodetection of TRPC5 in BAECs
Immunocytochemical staining of BAECs was done as described previously, with slight modifications [20]. The fixed cells were incubated with or without primary antibodies of rabbit anti-TRPC5 (dilution 1:100, Alomone Lab) and goat antiplatelet endothelial cell adhesion molecule 1 (anti-PECAM1, dilution 1:100, Santa Cruz) at 4°C overnight, followed by incubation with secondary donkey anti-rabbit IgG conjugated to Alexa Fluor 488 (1:200) and donkey anti-goat IgG conjugated to Alexa Fluor 546 (1:200). Immunofluorescence was detected by FV1000 confocal system (Olympus). Fluorescence signals were acquired at the settings under which the signals from the control cells (without primary antibodies) were absent.
Anti-TRPC5 blocking antibody
Anti-TRPC5-blocking antibody (T5E3) was raised in rabbits by replicating a previously described strategy [22]. Briefly, a peptide corresponding to TRPC5 putative pore region (CYETRAIDEPNNCKG) was synthesized and conjugated to keyhole limpet hemocyanin at Alpha Diagnostic International. The conjugated T5E3 peptide was injected subcutaneously to the back of a rabbit followed by two booster doses. T5E3 antiserum was collected 4 weeks after the second booster. IgG was purified from T5E3 antiserum and preimmune serum using a protein G column. Antibodies were applied to the cells at 4 µg/ml.
Chemicals and reagents
Carbachol (Calbiochem), LaCl3 (Sigma), and adenosine triphosphate sodium salt (ATP; Sigma) were prepared as 100 mM stock solutions in H2O. 1-oleoyl-2-acetyl-sn-glycerol (OAG; Calbiochem), KT5823 (Calbiochem), ODQ (Calbiochem), 8-bromo-cGMP (Calbiochem), and 2-aminoethoxydiphenyl borate (2-APB; Calbiochem) solution were prepared in DMSO. SNAP (Sigma) was prepared as a 300- or 250-mM stock dissolved in DMSO, and the aliquots were stored at −20°C. DEA-NONOate (Cayman) was prepared as a 100- or 30-mM stock dissolved in ethanol or DMSO, and the aliquots were stored at −80°C.
Data analysis and presentation
Data from Ca2+-imaging were plotted as traces and expressed as mean ± SEM of cells/wells in one experiment. Traces from patch-clamp recording and arterial tension measurement are representative individual experiments. Bar charts show data in mean ± SEM of all or combined experiments. Number of individual recordings “n” is given in parenthesis. Student’s t test was used for statistical comparison, with probability p < 0.05 (single asterisk) or p < 0.01 (double asterisks) as a significant difference.
Results
Lack of effect of NO donors on Ca2+-influx mediated by TRPC5
In single-cell intracellular Ca2+ measurement studies of mTRPC5, cells were exposed to a high concentration of SNAP (300 µM). In 18 out of 23 independent experiments, SNAP failed to elicit a significant response (e.g., Fig. 1a). In the other five experiments, SNAP elicited a small rise in [Ca2+]i but this effect was not unique to the TRPC5-expressing cells, occurring also in the vector control group (Suppl. Fig. 2). Addition of carbachol (CCh), which acts as an agonist at endogenous muscarinic receptors, caused marked elevation of [Ca2+]i in TRPC5-expressing cells (Fig. 1a) due to IP3-sensitive store Ca2+ release and receptor-mediated activation of TRPC5 [14]. Lanthanum (La3+), which stabilizes the open state of TRPC5 channels [10], enhanced the Ca2+ signal and 2-APB, which blocks TRPC5 channels [21], suppressed the signal (Fig. 1a). In vector-transfected cells, CCh elicited a small transient rise in [Ca2+]i, which is due to TRPC5-independent Ca2+-release, and La3+ had no effect (Fig. 1b, c). Analysis of data from all experiments indicated no significant difference between SNAP responses in TRPC5- and vector-transfected cells (Fig. 1c). The data suggest that mTRPC5 is stimulated by CCh and La3+ but not by SNAP.

SNAP on TRPC5-dependent Ca2+-entry in HEK293 cells. a–c mTRPC5 single-cell Ca2+ data. a, b Representative time-series graphs for [Ca2+]i in vector-transfected (b) and mTRPC5-transfected HEK293 cells (a). The bath solution contained NPSS. Arrows indicate the time points when chemicals were added (SNAP, 300 µM; carbachol 100 µM; LaCl3 100 µM; 2-APB, 75 µM). Each trace represents the mean ± SEM for ≥20 cells. c Mean ± SEM data for the [Ca2+]i response 1 min after application of CCh or the maximal [Ca2+]i response to La3+ and SNAP, as illustrated in a and b. Each bar represents the mean value from seven independent experiments. d–f hTRPC5 multiwell Ca2+ data. Shown are representative time-series graphs for [Ca2+]i in response to SNAP (100 µM, 500 µM), DMSO (0.2%), and Gd3+ (100 µM) in HEK293 cells stably expressing hTRPC5 under a tetracycline-inducible promoter with (Tet+; d) and without tetracycline (Tet−; e) induction. The data points represent the mean for ≥ 3 wells (SEM bars have been omitted for clarity; for full data sets see Suppl. Fig. 3). f Mean ± SEM (n = 3) data for the maximal [Ca2+]i change evoked by the applied chemicals
Multiwell intracellular Ca2+ measurement studies of hTRPC5 revealed similar insensitivity to SNAP, with 100 μM SNAP eliciting no rise in [Ca2+]i. High concentrations of SNAP (250 or 500 μM) elicited a [Ca2+]i rise in some batches of hTRPC5-expressing cells but these concentrations also had effects in control cells (Fig. 1d, e). Comprehensive presentation of all individual data sets is shown in Suppl. Fig. 3, indicating that there was modest hTRPC5 stimulation by 250 or 500 μM SNAP in some recordings. However, statistical analysis of all data did not support a conclusion that hTRPC5 could be stimulated by any SNAP concentration (1–500 μM) on a reliable basis (Fig. 1f). Strong [Ca2+]i responses on addition of the lanthanide gadolinium (Gd3+) confirmed that there was functional expression of TRPC5 in Tet+ but not Tet− (control) cells (Fig. 1d–f). Concentration–response data for Gd3+ established that the recording technique conferred the expected sensitivity (data not shown).
A chemically distinct NO donor, DEA-NONOate, was also tested using the single-cell (mTRPC5) and multiwell (hTRPC5) Ca2+ measurement systems. DEA-NONOate, at a range of concentrations from 10 to 100 µM, failed to elicit [Ca2+]i rise in mTRPC5-expressing cells (Fig. 2a, c). Similarly, 1 to 150 μM DEA-NONOate elicited no response in cells expressing hTRPC5 and responding to Gd3+ (Fig. 2b, c). Preincubation with 10–100 μM DEA-NONOate did not affect Gd3+ responses (Fig. 2d).

DEA-NONOate on TRPC5-dependent Ca2+-entry. a, b Representative time-series graphs for [Ca2+]i in mTRPC5-expressing HEK293 cells in response to DEA-NONOate (100 μM) (a the data points represents the mean ± SEM for ≥20 cells in single-cell Ca2+ measurement, final vehicle concentration of 0.1% DMSO) and in hTRPC5-expressing HEK293 cells (b the data points represent the mean ± SEM for ≥ 3 wells in multiwell Ca2+ measurement). c Mean ± SEM (n ≥ 3) data for the maximal [Ca2+]i change in response to DEA-NONOate as in a (n = 8) and b (n = 3). d Representative time-series graphs for [Ca2+]i in hTRPC5-expressing HEK293 cells in response to Gd3+ after DEA-NONOate. Carbachol (CCh), 100 µM; LaCl3 (La3+), 100 µM; GdCl3 (Gd3+), 100 µM; and DEA-NONOate (DEA), as indicated (μM); 1% DMSO in b and d
Lack of effect on ionic current mediated by TRPC5
Whole-cell currents were measured from hTRPC5-expressing HEK293 (Tet+) cells using a voltage-ramp protocol (Fig. 3). Spontaneously active TRPC5-mediated currents occurred, as shown by the example time-series relationship for +80 and −80 mV (Fig. 3a) and current–voltage relationship (I–V; Fig. 3b), which is typical of TRPC5 [14, 23, 25]. Bath application of 150 μM DEA-NONOate failed to stimulate or inhibit the current (Fig. 3a, b). In contrast, the reducing agents dithiothreitol or reduced thioredoxin elicited characteristic TRPC5 currents (Fig. 3a–d), as reported previously [23]. Neither DEA-NONOate nor SNAP had significant effects on the TRPC5 currents (Fig. 3d). The data further suggest that TRPC5 is resistant to NO.

Ionic current through TRPC5 channels during whole-cell voltage clamp. Recordings were from HEK293 cells expressing hTRPC5. a Representative time course of whole-cell current sampled at +80 mV and −80 mV during a voltage ramp and showing bath application of 150 µM DEA-NONOate and 10 mM DTT. b, c Current–voltage relationships (I–Vs) for the experiment in a showing the initial (basal) current and the current in the presence of DEA-NONOate (b), and the current in the presence of DEA-NONOate and the current induced by DTT (c). d Mean change (▵) in inward current at −80 mV after application of 150 µM DEA-NONOate, 10 mM DTT, 500 µM SNAP (10 min) or 4 µg/ml rTRX (5 min; n ≥ 3)
Effectiveness of NO donors to deliver NO in experiments
As positive controls for the NO donors, we first performed isometric contraction studies on mouse aortic segments where NO donors are known to act as potent vasorelaxants [9]. SNAP elicited relaxation of tone at 0.1–1 μM concentrations, while DEA-NONOate elicited relaxations at 10 and 100 μM concentrations (Fig. 4a, b). Treatment of HEK293 cells with these two NO donors also caused substantial elevation of intracellular NO as monitored by the NO-sensitive fluorescence dye DAF-FM acetate (Fig. 4c–f). As shown, extracellular application of 100 μM SNAP (Fig. 4c, e) or 90 μM DEA-NONOate (Fig. 4d, f) increased the intracellular NO level in a time-dependent manner. We also tested the biological activity of SNAP and DEA-NONOate through their expected inhibitory effects on TRPC6. TRPC6 and TRPC3 are two TRPC isoforms that were previously reported to be inhibited by NO-cGMP-PKG pathway [12, 18]. Application of the TRPC6 activator OAG induced a [Ca2+]i rise in TRPC6-expressing HEK293 cells (Fig. 4g). This [Ca2+]i rise was attenuated by a PKG activator 8-bromo-cGMP and enhanced by a PKG inhibitor KT5823 or ODQ, a guanylate cyclase inhibitor (Fig. 4h). Importantly, 300 μM SNAP or 100 μM DEA-NONOate attenuated the OAG-induced [Ca2+]i rise in these cells (Fig. 4g, h). The inhibitory effect of DEA-NONOate was reversed by KT5823 (Fig. 4g, h). These results confirmed that SNAP and DEA-NONOate applied in our systems could effectively release NO to inhibit TRPC6 via NO-cGMP-PKG pathway [12, 18].
Collectively, these experiments demonstrate that the NO donors used in our experiments were able to efficiently deliver biologically active NO.

Validation of NO-releasing capacities of SNAP and DEA-NONOate. a, b Representative time courses of isometric tension in isolated mouse aortic segments in response to cumulatively increasing concentrations of SNAP (a) and DEA-NONOate (b) applied to the bath. The vessels were denuded of the endothelial layer and preconstricted with 10 μM phenylephrine (Phe; n = 4). c, d Representative time-series graphs for change of DAF-FM fluorescence (ΔF*) in HEK293 cells in response to SNAP (100 μM, c) or DEA-NONOate (90 μM, d). Also shown are responses to vehicle (DMSO, 0.2% in c; DMSO, 1.5% in d). Each data point represents mean ± SEM for measurements from four wells of a 96-well plate. e, f For three independent experiments, mean ± SEM data for the maximal ΔF* in response to SNAP (e) or DEA-NONOate (f) as in c and d. g Representative time-series traces (n ≥ 6) for [Ca2+]i in response to 100 μM OAG in HEK293 cells cotransfected with PKG1α and mTRPC6. Prior to OAG addition, cells were pretreated for 5 min with 0.1% DMSO, 300 μM SNAP, 100 μM DEA-NONOate (DEA), or 100 μM DEA-NONOate plus 1 μM KT5823 (DEA+KT5823). The data points represent the mean ± SEM for ≥20 cells in a single experiment. h Summary of data showing maximal [Ca2+]i response (Fmax/F0) to OAG (100 μM) challenge under different pretreatments for 5 min. DMSO, 0.1%; KT5823, 1 μM; ODQ, 50 μM; 8-bromo-cGMP, 2 mM; SNAP, 300 μM; and DEA-NONOate (DEA), 100 μM. Mean ± SEM (n ≥ 6 experiments)
Inhibition of Ca2+-entry in endothelial cells (BAECs)
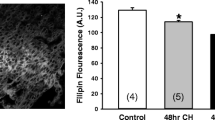
Resistance of TRPC5 to NO in our experiments but sensitivity in studies of other investigators [24] may have arisen because NO does not act directly but requires an intermediate protein that is inadequately expressed in our HEK293 cells relative to TRPC5. The identity of such an intermediate is unknown and so we sought to address the possibility by studying an endogenous Ca2+-entry signal that involves TRPC5. The previous reports suggested that TRPC5 is expressed in BAECs and is functionally important in agonist-stimulated Ca2+-entry and spontaneous Ca2+-entry in these cells [20, 24]. Figure 5a showed that TRPC5 immunoreactive signals could be detected in PECAM-1 (an endothelial cell marker)-positive BAECs. Many research groups have investigated the effect of NO on endothelial cell [Ca2+]i signaling, While most reports showed an inhibitory action of NO on [Ca2+]i in endothelial cells [5, 11, 18, 21], there were also a few reports showing stimulatory action of NO donor SNAP on [Ca2+]i in endothelial cells [13, 24]. In our studies, direct application of 10 and 300 μM SNAP to cultured BAECs did not cause significant change in [Ca2+]i level (n = 4).

Endogenous TRPC5 expression in BAECs and inhibitory effect of SNAP on ATP-stimulated Ca2+-entry involving TRPC5. a Immunocytochemical detection of TRPC5 in BAECs. The cells were stained with primary antibodies targeted to TRPC5 (TRPC5; green signals) and to platelet endothelial cell adhesion molecule 1 (PECAM1; red signals). Also shown were the merged fluorescent signals overlaid on the bright-view image (Merged). The control image was the merged image from sample with the omission of primary antibodies (Control). b Inhibitory effect of T5E3 on LaCl3 (La3+, 100 µM) and carbachol (CCh, 100 µM)-induced [Ca2+]i rises in mTRPC5-over-expressing HEK293 cells with or without 4 µg/ml anti-TRPC5 blocking antibody (T5E3) in the bath solution. Mean ± SEM (n = 3) data are shown (*p < 0.05). Representative experimental traces are given in Supplementary Fig. 4. c OAG (100 µM)-induced [Ca2+]i rises in TRPC3-over-expressing HEK293 cells with 4 µg/ml preimmune IgG or T5E3 in the bath solution. Mean ± SEM (n = 3) data are shown (*p < 0.05). Representative experimental traces are given in Supplementary Fig. 4. d, e Representative time courses of [Ca2+]i changes in BAECs in response to ATP (100 µM). BAECs were bathed in HBS containing 4 µg/ml T5E3 (upper trace of e) or preimmune IgG (d, lower trace of e) for 20–30 min at room temperature prior to Ca2+ imaging. Bath solution was changed to HBS-EGTA containing preimmune IgG or T5E3 at the beginning of each recording. 300 µM SNAP (lower trace of e) or 0.1% DMSO (d, upper trace of e) was added to the bath, followed by addition of ATP to elicit store-release. After ATP-addition (8 min), the Ca2+ influx was initiated by changing the bath solution to HBS containing ATP, DMSO/SNAP with T5E3/preimmune IgG. Each trace represents the mean ± SEM of ≥10 cells in one representative experiment. f Summary of data for all independent experiments of the type shown in d and e, showing the maximal [Ca2+]i change after restoring Ca2+ in bath (n = 6–10; **p < 0.01)
We then used a TRPC5-specific blocking antibody T5E3 [22] to investigate the TRPC5 component in the agonist-stimulated Ca2+-entry and spontaneous Ca2+-entry. Effectiveness of T5E3 as an inhibitor of TRPC5 was confirmed in HEK293 cells over-expressing mTRPC5 (Fig. 5b and Suppl. Fig. 4). T5E3 inhibited the La3+ and carbachol-stimulated Ca2+ influx in these cells (Fig. 5b and Suppl. Fig. 4). Specificity of T5E3 was shown through lack of effect of T5E3 on OAG-stimulated [Ca2+]i rises in HEK293 cells over-expressing TRPC3 (Fig. 5c and Suppl. Fig. 4).
ATP stimulates TRPC5 activity via a receptor-operated pathway in BAECs and other cell types [1, 24]. We exposed the BAECs to anti-TRPC5 blocking antibody, T5E3, or its control preimmune antibody (Fig. 5d–f). T5E3 suppressed the ATP-stimulated Ca2+-entry (Fig. 5d–f) and ATP-stimulated whole-cell current (Suppl. Fig. 6), consistent with TRPC5 having a role in the response. The NO donor SNAP was tested against the Ca2+-entry and was found to be inhibitory (Fig. 5d–f). Furthermore, in T5E3-treated cells, SNAP generated no further inhibition (column 2 and 4 in Fig. 5f); in addition, T5E3 had no further inhibition in the presence of SNAP (column 3 and 4 in Fig. 5f), suggesting that SNAP and T5E3 inhibited the same component.
To study spontaneous Ca2+-entry in BAECs, we first exposed the BAECs to Ca2+-free physiological medium, and then reintroduced 2 mM extracellular Ca2+. Ca2+-entry was observed (Fig. 6a, c). This Ca2+-entry was again suppressed by T5E3 and SNAP (Fig. 6a–c). We also used a dominant-negative TRPC5 construct (DN-T5) that can disrupt TRPC5 function [17]. Expression of DN-T5 also reduced the spontaneous Ca2+-entry in BAECs (Fig. 6d–f), supporting involvement of channels that can incorporate TRPC5. Moreover, SNAP inhibited the same component as T5E3 and DN-T5 did (Fig. 6c, f). Taken together, the data suggest that an endogenous Ca2+-entry mechanism involving TRPC5 in endothelial cells is sensitive to inhibition by NO.

Inhibitory effect of SNAP on spontaneous Ca2+-entry involving TRPC5 in BAECs. a, b, d, e Representative time courses of [Ca2+]i changes in BAECs in response to extracellular Ca2+ readdition. a, b Cells were incubated in HBS containing 4 µg/ml T5E3 (upper trace of b) or preimmune IgG (a, lower trace of b) for 20–30 min prior to Ca2+ imaging. d, e Cells were transfected with empty expression vector (vector) or DN-T5 construct (DN-T5). 300 µM SNAP (lower trace of b and e) or 0.1% DMSO (a and d, upper trace of b and e) was added to the bath for 15 min, followed by changing the bath solution to HBS DMSO/SNAP with T5E3/preimmune IgG. Each trace represents the mean ± SEM of ≥5 cells in one representative experiment. c, f Summary of data for all independent experiments of the type shown in a, b (c), d, e (f), showing the maximal [Ca2+]i change after restoring Ca2+ in bath. Mean ± SEM (n = 4 for c; n = 5–10 for f) data are shown (*p < 0.05; **p < 0.01)
Discussion
In the present study, we found that NO donors fail to modulate activity of mTRPC5 or hTRPC5 channels exogenously expressed in HEK293 cells when applied at concentrations that are sufficient for generation of NO, evoke vascular relaxation, or suppress TRPC6 via protein kinase G. We did, however, find an inhibitory effect on an endothelial Ca2+-entry mechanism that involved TRPC5. The data suggest that NO is not a direct modulator of TRPC5 but that it can negatively modulate endogenous TRPC5-containing channels through an indirect mechanism.
We occasionally observed a stimulatory effect of SNAP that appeared greater than the response in control cells (Fig. 1f, Suppl. Fig. 3). Although these latter results did not achieve statistical significance, underlying examples (Suppl. Fig. 3) are reminiscent of the results of Yoshida et al. [24]. Therefore, we believe that TRPC5 may be stimulated by SNAP under some conditions but we suggest that any such effect is not due to an action of nitric oxide and occurs only at higher concentrations of SNAP than are necessary to evoke nitric oxide-dependent effects. High concentrations of SNAP may generate free radicals [19], which Yoshida et al. [24] also suggested activate TRPC5. We did not observe any effects of DEA-NONOate.
One concern might be the stability of NO donors. The question is whether SNAP and DEA-NONOate applied to the cells could indeed increase NO concentration in cultured cells. In this regard, strict protocols were followed to preserve the activity of the NO donors. The same NO donor preparations relaxed aortic strips concentration-dependently, elicited NO rises in HEK293 cells, and inhibited TRPC6 via a cGMP-PKG pathway (Fig. 4). The data substantiate the competence of the NO donors in our experimental settings.
The data in our study further support the conclusion that the cysteine residues in the ion pore region of TRPC5 (cysteines 553 and 558) are involved in an extracellular disulfide bridge [23]. Such covalent bonding precludes availability of the cysteine residues for S-nitrosylation unless the bridge is broken by a reducing agent. Consistent with this conclusion, mutating the cysteines resulted in fully active channels that were unresponsive to lanthanide [23].
The observed inhibitory effect on an endothelial Ca2+-entry mechanism suggests the possibility for an effect of NO on a Ca2+ channel complex involving TRPC5. Such an inhibitory effect may have physiological importance as a negative feedback mechanism in the control of NO release. However, failure to observe stimulation or inhibition of over-expressed TRPC5 by NO suggests that the endothelial effect did not occur through a direct action of NO on TRPC5.
Our results in TRPC5-over-expressing HEK293 cells and BAECs show some divergence from those of Yoshida et al. and others [13, 24]. In additional experiments, we used the same composition of extracellular solution and TRPC5 clone as Yoshida et al. [24] but were unable to observe a stimulatory effect (Suppl. Fig. 5). The reason for the discrepancy in results is not clear. Possible complicating factors could include differences in HEK293 cell sources, BAEC isolation procedures, and cell culture conditions. Such factors may alter the expression or activity of certain cellular factors that may be required for an indirect action of SNAP. Another possibility is that a stimulating effect of SNAP on basal [Ca2+]i of some HEK293 cells might enhance TRPC5 activity via Ca2+-dependent potentiation.
In summary, the study provides evidence against NO as a direct modulator of TRPC5 channels and against S-nitrosylation as a mechanism for stimulation of TRPC5 channels in the absence of a reducing factor. The study does, however, suggest that NO may indirectly inhibit endogenous Ca2+-entry mechanisms involving TRPC5.
References
Beech DJ (2007) Canonical transient receptor potential 5. Handb Exp Pharmacol 176:109–123
Bicker G (2005) STOP and GO with NO: nitric oxide as a regulator of cell motility in simple brains. Bioessays 27:495–505
Cheng KT, Leung YK, Shen B, Kwok YC, Wong CO, Kwan HY, Man YB, Ma X, Huang Y, Yao X (2008) CNGA2 channels mediate adenosine-induced Ca2+ influx in vascular endothelial cells. Arterioscler Thromb Vasc Biol 28:913–918
Chung HT, Choi BM, Kwon YG, Kim YM (2008) Interactive relations between nitric oxide (NO) and carbon monoxide (CO): heme oxygenase-1/CO pathway is a key modulator in NO-mediated antiapoptosis and anti-inflammation. Methods Enzymol 441:329–338
Dedkova EN, Blatter LA (2002) Nitric oxide inhibits capacitative Ca2+ entry and enhances endoplasmic reticulum Ca2+ uptake in bovine vascular endothelial cells. J Physiol 539:77–91
Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS (2005) Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6:150–166
Ignarro LJ (2002) Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol 53:503–514
Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 3:193–197
Jiang F, Gibson AP, Dusting GJ (2001) Endothelial dysfunction induced by oxidized low-density lipoproteins in isolated mouse aorta: a comparison with apolipoprotein-E deficient mice. Eur J Pharmacol 424:141–149
Jung S, Muhle A, Schaefer M, Strotmann R, Schultz G, Plant TD (2003) Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J Biol Chem 278:3562–3571
Kwan HY, Huang Y, Yao X (2000) Store-operated calcium entry in vascular endothelial cells is inhibited by cGMP via a protein kinase G-dependent mechanism. J Biol Chem 275:6758–6763
Kwan HY, Huang Y, Yao X (2004) Regulation of canonical transient receptor potential isoform 3 (TRPC3) channel by protein kinase G. Proc Natl Acad Sci USA 101:2625–2630
Mottola A, Antoniotti S, Lovisolo D, Munaron L (2005) Regulation of noncapacitative calcium entry by arachidonic acid and nitric oxide in endothelial cells. FASEB J 19:2075–2077
Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G (2000) Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem 275:17517–17526
Snyder SH (1994) Nitric oxide. More jobs for that molecule. Nature 372:504–505
Stamler JS, Toone EJ, Lipton SA, Sucher NJ (1997) (S)NO signals: translocation, regulation, and a consensus motif. Neuron 18:691–696
Strubing C, Krapivinsky G, Krapivinsky L, Clapham DE (2003) Formation of novel TRPC channels by complex subunit interactions in embryonic brain. J Biol Chem 278:39014–39019
Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R (2008) Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol 586:4209–4223
Wei T, Chen C, Hou J, Xin W, Mori A (2000) Nitric oxide induces oxidative stress and apoptosis in neuronal cells. Biochim Biophys Acta 1498:72–79
Wong CO, Huang Y, Yao X (2010) Genistein potentiates activity of the cation channel TRPC5 independently of tyrosine kinases. Br J Pharmacol 159:1486–1496
Xu SZ, Zeng F, Boulay G, Grimm C, Harteneck C, Beech DJ (2005) Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br J Pharmacol 145:405–414
Xu SZ, Boulay G, Flemming R, Beech DJ (2006) E3-targeted anti-TRPC5 antibody inhibits store-operated calcium entry in freshly isolated pial arterioles. Am J Physiol Heart Circ Physiol 291:H2653–H2659
Xu SZ, Sukumar P, Zeng F, Li J, Jairaman A, English A, Naylor J, Ciurtin C, Majeed Y, Milligan CJ, Bahnasi YM, Al-Shawaf E, Porter KE, Jiang LH, Emery P, Sivaprasadarao A, Beech DJ (2008) TRPC channel activation by extracellular thioredoxin. Nature 451:69–72
Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, Tominaga M, Shimizu S, Sato Y, Mori Y (2006) Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol 2:596–607
Zeng F, Xu SZ, Jackson PK, McHugh D, Kumar B, Fountain SJ, Beech DJ (2004) Human TRPC5 channel activated by a multiplicity of signals in a single cell. J Physiol 559:739–750
Acknowledgments
We are grateful for support from Hong Kong RGC grant (CUHK477307, CUHK477408, and CUHK479109), Focused Investment Scheme of Chinese University of Hong Kong, and Li Ka Shing Institute of Health Sciences. DJB research was funded by the Wellcome Trust and Overseas Research and University scholarships to PS. We thank Y. Mori for providing mTRPC5 and SNAP and for comments on the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Ching-On Wong and Piruthivi Sukumar contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Suppl. Fig. 1
(GIFF 94 kb)
Suppl. Fig. 2
(GIFF 46 kb)
Suppl. Fig. 3-1
(GIFF 82 kb)
Suppl. Fig. 3-2
(GIFF 170 kb)
Suppl. Fig. 3-3
(GIFF 111 kb)
Suppl. Fig. 4
(GIFF 77 kb)
Suppl. Fig. 5
(GIFF 48 kb)
Suppl. Fig. 6
(GIFF 34 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wong, CO., Sukumar, P., Beech, D.J. et al. Nitric oxide lacks direct effect on TRPC5 channels but suppresses endogenous TRPC5-containing channels in endothelial cells. Pflugers Arch - Eur J Physiol 460, 121–130 (2010). https://doi.org/10.1007/s00424-010-0823-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-010-0823-3