
Abstract
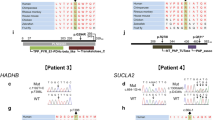
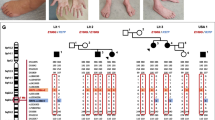
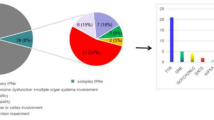
Inherited peripheral neuropathies (IPN) are one of the most frequent inherited causes of neurological disability characterized by considerable phenotypic and genetic heterogeneity. Based on clinical and electrophysiological properties, they can be subdivided into three main groups: HMSN, dHMN, and HSN. At present, more than 50 IPN genes have been identified. Still, many patients and families with IPN have not yet received a molecular genetic diagnosis because clinical genetic testing usually only covers a subset of IPN genes. Moreover, a considerable proportion of IPN genes has to be identified. Here we present results of WES in 27 IPN patients excluded for mutations in many known IPN genes. Eight of the patients received a definite diagnosis. While six of these patients carried bona fide pathogenic mutations in known IPN genes, two patients had mutations in genes known to be involved in other types of neuromuscular disorders. A further group of eight patients carried sequence variations in IPN genes that could not unequivocally be classified as pathogenic. In addition, combining data of WES and linkage analysis identified SH3BP4, ITPR3, and KLHL13 as novel IPN candidate genes. Moreover, there was evidence that particular mutations in PEX12, a gene known to cause Zellweger syndrome, could also lead to an IPN phenotype. We show that WES is a useful tool for diagnosing IPN and we suggest an expanded phenotypic spectrum of some genes involved in other neuromuscular and neurodegenerative disorders. Nevertheless, interpretation of variants in known and potential novel disease genes has remained challenging.
Similar content being viewed by others
References
Skre H (1974) Genetic and clinical aspects of Charcot–Marie–Tooth’s disease. Clin Genet 6:98–118
Dyck PJ, Chance P, Lebo R, Carney JA (1993) Hereditary motor and sensory neuropathies. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF (eds) Peripheral neuropathy, 3rd edn. Saunders, Philadelphia, pp 1094–1136
Reilly MM, Murphy SM, Laurá M (2011) Charcot–Marie–Tooth disease. J Peripher Nerv Syst 16:1–14
Harding AE (1993) Inherited neuronal atrophy and degeneration predominantly of lower motor neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF (eds) Peripheral neuropathy, 3rd edn. Saunders, Philadelphia, pp 1051–1064
Rossor AM, Kalmar B, Greensmith L, Reilly MM (2012) The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry 83:6–14
Antonellis A, Ellsworth RE, Sambuughin N, Puls I, Abel A, Lee-Lin SQ, Jordanova A, Kremensky I, Christodoulou K, Middleton LT, Sivakumar K, Ionasescu V, Funalot B, Vance JM, Goldfarb LG, Fischbeck KH, Green ED (2003) Glycyl tRNA synthetase mutations in Charcot–Marie–Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet 72:1293–1299
Auer-Grumbach M, Schlotter-Weigel B, Lochmüller H, Strobl-Wildemann G, Auer-Grumbach P, Fischer R, Offenbacher H, Zwick EB, Robl T, Hartl G, Hartung HP, Wagner K, Windpassinger C, Austrian Peripheral Neuropathy Study Group (2005) Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann Neurol 57:415–424
Dyck PJ (1993) Neuronal atrophy and degeneration predominantly affecting peripheral sensory and autonomic neurons. In: Dyck PJ, Thomas PK, Griffin JW, Low PA, Poduslo JF (eds) Peripheral neuropathy, 3rd edn. Saunders, Philadelphia, pp 1065–1093
Auer-Grumbach M, Mauko B, Auer-Grumbach P, Pieber TR (2006) Molecular genetics of hereditary sensory neuropathies. Neuromol Med 8:147–158
Kiwaki T, Umehara F, Takashima H, Nakagawa M, Kamimura K, Kashio N, Sakamoto Y, Unoki K, Nobuhara Y, Michizono K, Watanabe O, Arimura H, Osame M (2000) Hereditary motor and sensory neuropathy with myelin folding and juvenile onset glaucoma. Neurology 55:392–397
Münch C, Rosenbohm A, Sperfeld AD, Uttner I, Reske S, Krause BJ, Sedlmeier R, Meyer T, Hanemann CO, Stumm G, Ludolph AC (2005) Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol 58:777–780
Auer-Grumbach M, Weger M, Fink-Puches R, Papić L, Fröhlich E, Auer-Grumbach P, El Shabrawi-Caelen L, Schabhüttl M, Windpassinger C, Senderek J, Budka H, Trajanoski S, Janecke AR, Haas A, Metze D, Pieber TR, Guelly C (2011) Fibulin-5 mutations link inherited neuropathies, age-related macular degeneration and hyperelastic skin. Brain 134:1839–1852
Murphy SM, Laura M, Fawcett K, Pandraud A, Liu YT, Davidson GL, Rossor AM, Polke JM, Castleman V, Manji H, Lunn MP, Bull K, Ramdharry G, Davis M, Blake JC, Houlden H, Reilly MM (2012) Charcot–Marie–Tooth disease: frequency of genetic subtypes and guidelines for genetic testing. J Neurol Neurosurg Psychiatry 83:706–710
Lupski JR, Reid JG, Gonzaga-Jauregui C, Rio Deiros D, Chen DC, Nazareth L, Bainbridge M, Dinh H, Jing C, Wheeler DA, McGuire AL, Zhang F, Stankiewicz P, Halperin JJ, Yang C, Gehman C, Guo D, Irikat RK, Tom W, Fantin NJ, Muzny DM, Gibbs RA (2010) Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 362:1181–1191
Montenegro G, Powell E, Huang J, Speziani F, Edwards YJ, Beecham G, Hulme W, Siskind C, Vance J, Shy M, Züchner S (2011) Exome sequencing allows for rapid gene identification in a Charcot–Marie–Tooth family. Ann Neurol 69:464–470
Beetz C, Pieber TR, Hertel N, Schabhüttl M, Fischer C, Trajanoski S, Graf E, Keiner S, Kurth I, Wieland T, Varga RE, Timmerman V, Reilly MM, Strom TM, Auer-Grumbach M (2012) Exome sequencing identifies a REEP1 mutation involved in distal hereditary motor neuropathy type V. Am J Hum Genet 91:139–145
Choi BO, Koo SK, Park MH, Rhee H, Yang SJ, Choi KG, Jung SC, Kim HS, Hyun YS, Nakhro K, Lee HJ, Woo HM, Chung KW (2012) Exome sequencing is an efficient tool for genetic screening of Charcot–Marie–Tooth disease. Hum Mutat 33:1610–1615
Zimoń M, Baets J, Almeida-Souza L, De Vriendt E, Nikodinovic J, Parman Y, Battaloğlu E, Matur Z, Guergueltcheva V, Tournev I, Auer-Grumbach M, De Rijk P, Petersen BS, Müller T, Fransen E, Van Damme P, Löscher WN, Barišić N, Mitrovic Z, Previtali SC, Topaloğlu H, Bernert G, Beleza-Meireles A, Todorovic S, Savic-Pavicevic D, Ishpekova B, Lechner S, Peeters K, Ooms T, Hahn AF, Züchner S, Timmerman V, Van Dijck P, Rasic VM, Janecke AR, De Jonghe P, Jordanova A (2012) Loss-of-function mutations in HINT1 cause axonal neuropathy with neuromyotonia. Nat Genet 44:1080–1083
Auer-Grumbach M, Strasser-Fuchs S, Robl T, Windpassinger C, Wagner K (2003) Late onset Charcot–Marie–Tooth 2 syndrome caused by two novel mutations in the MPZ gene. Neurology 61:1435–1437
Rüschendorf F, Nürnberg P (2005) ALOHOMORA: a tool for linkage analysis using 10K SNP array data. Bioinformatics 21:2123–2125
Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101
Moser HW, Moser AB (1991) Measurement of saturated very long chain fatty acids in plasma. In: Hommes F (ed) Techniques in diagnostic human biochemical genetics. Wiley, New York, pp 177–191
Vreken P, van Lint AE, Bootsma AH, Overmars H, Wanders RJ, van Gennip AH (1998) Rapid stable isotope dilution analysis of very-long-chain fatty acids, pristanic acid and phytanic acid using gas chromatography-electron impact mass spectrometry. J Chromatogr B Biomed Sci Appl 713:281–287
Auer-Grumbach M, Bode H, Pieber TR, Schabhüttl M, Fischer D, Seidl R, Graf E, Wieland T, Schuh R, Vacariu G, Grill F, Timmerman V, Strom TM, Hornemann T (2013) Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur J Med Genet 56:266–269
Sivakumar K, Kyriakides T, Puls I, Nicholson GA, Funalot B, Antonellis A, Sambuughin N, Christodoulou K, Beggs JL, Zamba-Papanicolaou E, Ionasescu V, Dalakas MC, Green ED, Fischbeck KH, Goldfarb LG (2005) Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain 128:2304–2314
Cali JJ, Hsieh CL, Francke U, Russell DW (1991) Mutations in the bile acid biosynthetic enzyme sterol 27-hydroxylase underlie cerebrotendinous xanthomatosis. J Biol Chem 266:7779–7783
Guyant-Maréchal L, Verrips A, Girard C, Wevers RA, Zijlstra F, Sistermans E, Vera P, Campion D, Hannequin D (2005) Unusual cerebrotendinous xanthomatosis with fronto-temporal dementia phenotype. Am J Med Genet A 139A:114–117
Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33:455–456
Münch C, Sedlmeier R, Meyer T, Homberg V, Sperfeld AD, Kurt A, Prudlo J, Peraus G, Hanemann CO, Stumm G, Ludolph AC (2004) Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 63:724–726
Verhoeven K, De Jonghe P, Van de Putte T, Nelis E, Zwijsen A, Verpoorten N, De Vriendt E, Jacobs A, Van Gerwen V, Francis A, Ceuterick C, Huylebroeck D, Timmerman V (2003) Slowed conduction and thin myelination of peripheral nerves associated with mutant rho Guanine-nucleotide exchange factor 10. Am J Hum Genet 73:926–932
Jordanova A, Irobi J, Thomas FP, Van Dijck P, Meerschaert K, Dewil M, Dierick I, Jacobs A, De Vriendt E, Guergueltcheva V, Rao CV, Tournev I, Gondim FA, D’Hooghe M, Van Gerwen V, Callaerts P, Van Den Bosch L, Timmermans JP, Robberecht W, Gettemans J, Thevelein JM, De Jonghe P, Kremensky I, Timmerman V (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot–Marie–Tooth neuropathy. Nat Genet 38:197–202
Latour P, Thauvin-Robinet C, Baudelet-Méry C, Soichot P, Cusin V, Faivre L, Locatelli MC, Mayençon M, Sarcey A, Broussolle E, Camu W, David A, Rousson R (2010) A major determinant for binding and aminoacylation of tRNA(Ala) in cytoplasmic Alanyl-tRNA synthetase is mutated in dominant axonal Charcot–Marie–Tooth disease. Am J Hum Genet 86:77–82
Blumen SC, Astord S, Robin V, Vignaud L, Toumi N, Cieslik A, Achiron A, Carasso RL, Gurevich M, Braverman I, Blumen N, Munich A, Barkats M, Viollet L (2012) A rare recessive distal hereditary motor neuropathy with HSJ1 chaperone mutation. Ann Neurol 71:509–519
Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, De Jonghe P, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battaloglu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot–Marie–Tooth neuropathy type 2A. Nat Genet 36:449–451
Chung KW, Kim SB, Park KD, Choi KG, Lee JH, Eun HW, Suh JS, Hwang JH, Kim WK, Seo BC, Kim SH, Son IH, Kim SM, Sunwoo IN, Choi BO (2006) Early onset severe and late-onset mild Charcot–Marie–Tooth disease with mitofusin 2 (MFN2) mutations. Brain 129:2103–2118
Nicholson GA, Magdelaine C, Zhu D, Grew S, Ryan MM, Sturtz F, Vallat JM, Ouvrier RA (2008) Severe early-onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology 70:1678–1681
Polke JM, Laurá M, Pareyson D, Taroni F, Milani M, Bergamin G, Gibbons VS, Houlden H, Chamley SC, Blake J, Devile C, Sandford R, Sweeney MG, Davis MB, Reilly MM (2011) Recessive axonal Charcot–Marie–Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology 77:168–173
Guilbot A, Williams A, Ravisé N, Verny C, Brice A, Sherman DL, Brophy PJ, LeGuern E, Delague V, Bareil C, Mégarbané A, Claustres M (2001) A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot–Marie–Tooth disease. Hum Mol Genet 10:415–421
Chang CC, Lee WH, Moser H, Valle D, Gould SJ (1997) Isolation of the human PEX12 gene, mutated in group 3 of the peroxisome biogenesis disorders. Nat Genet 15:385–388
Okumoto K, Shimozawa N, Kawai A, Tamura S, Tsukamoto T, Osumi T, Moser H, Wanders RJ, Suzuki Y, Kondo N, Fujiki Y (1998) PEX12, the pathogenic gene of group III Zellweger syndrome: cDNA cloning by functional complementation on a CHO cell mutant, patient analysis, and characterization of PEX12p. Mol Cell Biol 18:4324–4336
Gootjes J, Skovby F, Christensen E, Wanders RJ, Ferdinandusse S (2004) Reinvestigation of trihydroxycholestanoic acidemia reveals a peroxisome biogenesis disorder. Neurology 62:2077–2081
Robinson R, Carpenter D, Shaw MA, Halsall J, Hopkins P (2006) Mutations in RYR1 in malignant hyperthermia and central core disease. Hum Mutat 27:977–989
Martiniuk F, Mehler M, Pellicer A, Tzall S, La Badie G, Hobart C, Ellenbogen A, Hirschhorn R (1986) Isolation of a cDNA for human acid alpha-glucosidase and detection of genetic heterogeneity for mRNA in three alpha-glucosidase-deficient patients. Proc Natl Acad Sci USA 83:9641–9644
Vorgerd M, Burwinkel B, Reichmann H, Malin JP, Kilimann MW (1998) Adult-onset glycogen storage disease type II: phenotypic and allelic heterogeneity in German patients. Neurogenetics 1:205–211
Fleischman RA (1992) Human piebald trait resulting from a dominant negative mutant allele of the c-kit membrane receptor gene. J Clin Invest 89:1713–1717
Nalini A, Gayathri N, Yasha TC, Ravishankar S, Urtizberea A, Huehne K, Rautenstrauss B (2008) Clinical, pathological and molecular findings in two siblings with giant axonal neuropathy (GAN): report from India. Eur J Med Genet 51:426–435
Tazir M, Nouioua S, Magy L, Huehne K, Assami S, Urtizberea A, Grid D, Hamadouche T, Rautenstrauss B, Vallat JM (2009) Phenotypic variability in giant axonal neuropathy. Neuromuscul Disord 19:270–274
Buysse K, Vergult S, Mussche S, Ceuterick-de Groote C, Speleman F, Menten B, Lissens W, Van Coster R (2010) Giant axonal neuropathy caused by compound heterozygosity for a maternally inherited microdeletion and a paternal mutation within the GAN gene. Am J Med Genet A 152A:2802–2804
Azzedine H, Bolino A, Taïeb T, Birouk N, Di Duca M, Bouhouche A, Benamou S, Mrabet A, Hammadouche T, Chkili T, Gouider R, Ravazzolo R, Brice A, Laporte J, LeGuern E (2003) Mutations in MTMR13, a new pseudophosphatase homologue of MTMR2 and Sbf1, in two families with an autosomal recessive demyelinating form of Charcot–Marie–Tooth disease associated with early-onset glaucoma. Am J Hum Genet 72:1141–1153
Senderek J, Bergmann C, Weber S, Ketelsen UP, Schorle H, Rudnik-Schöneborn S, Büttner R, Buchheim E, Zerres K (2003) Mutation of the SBF2 gene, encoding a novel member of the myotubularin family, in Charcot–Marie–Tooth neuropathy type 4B2/11p15. Hum Mol Genet 12:349–356
Hirano R, Takashima H, Umehara F, Arimura H, Michizono K, Okamoto Y, Nakagawa M, Boerkoel CF, Lupski JR, Osame M, Arimura K (2004) SET binding factor 2 (SBF2) mutation causes CMT4B with juvenile onset glaucoma. Neurology 63:577–580
Zimoń M, Baets J, Auer-Grumbach M, Berciano J, Garcia A, Lopez-Laso E, Merlini L, Hilton-Jones D, McEntagart M, Crosby AH, Barisic N, Boltshauser E, Shaw CE, Landouré G, Ludlow CL, Gaudet R, Houlden H, Reilly MM, Fischbeck KH, Sumner CJ, Timmerman V, Jordanova A, De Jonghe P (2010) Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies. Brain 133:1798–1809
Sevilla T, Cuesta A, Chumillas MJ, Mayordomo F, Pedrola L, Palau F, Vílchez JJ (2003) Clinical, electrophysiological and morphological findings of Charcot–Marie–Tooth neuropathy with vocal cord palsy and mutations in the GDAP1 gene. Brain 126:2023–2033
Zimoń M, Baets J, Fabrizi GM, Jaakkola E, Kabzińska D, Pilch J, Schindler AB, Cornblath DR, Fischbeck KH, Auer-Grumbach M, Guelly C, Huber N, De Vriendt E, Timmerman V, Suter U, Hausmanowa-Petrusewicz I, Niemann A, Kochański A, De Jonghe P, Jordanova A (2011) Dominant GDAP1 mutations cause predominantly mild CMT phenotypes. Neurology 77:540–548
Tebar F, Sorkina T, Sorkin A, Ericsson M, Kirchhausen T (1996) Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J Biol Chem 271:28727–28730
Sidiropoulos PN, Miehe M, Bock T, Tinelli E, Oertli CI, Kuner R, Meijer D, Wollscheid B, Niemann A, Suter U (2012) Dynamin 2 mutations in Charcot–Marie–Tooth neuropathy highlight the importance of clathrin-mediated endocytosis in myelination. Brain 135:1395–1411
Maranto AR (1994) Primary structure, ligand binding, and localization of the human type 3 inositol 1,4,5-trisphosphate receptor expressed in intestinal epithelium. J Biol Chem 269:1222–1230
Toews JC, Schram V, Weerth SH, Mignery GA, Russell JT (2007) Signaling proteins in the axoglial apparatus of sciatic nerve nodes of Ranvier. Glia 55:202–213
Sumara I, Quadroni M, Frei C, Olma MH, Sumara G, Ricci R, Peter M (2007) A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell 12:887–900
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Acknowledgments
We are grateful for the participation of the patients and families in this study. This work was supported by the Austrian Science Fund (FWF, P23223-B19), the University of Antwerp (UA), the Association Belge contre les Maladies Neuromusculaires (ABMM), the Medical Foundation Queen Elisabeth (GSKE), the agency for Innovation by Science and Technology (IWT), the Fund for Scientific Research Flanders (FWO-Flanders) and the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement number 2012-305121 “Integrated European–omics research project for diagnosis and therapy in rare neuromuscular and neurodegenerative diseases (NEUROMICS)”.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Schabhüttl, M., Wieland, T., Senderek, J. et al. Whole-exome sequencing in patients with inherited neuropathies: outcome and challenges. J Neurol 261, 970–982 (2014). https://doi.org/10.1007/s00415-014-7289-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7289-8