
Abstract
GLUT1 deficiency syndrome (GLUT1DS) is a treatable neurometabolic disorder in which glucose transport into the brain is disturbed. Besides the classic phenotype of intellectual disability, epilepsy, and movement disorders, other phenotypes are increasingly recognized. These include, for example, idiopathic generalized epilepsy and paroxysmal exercise-induced dyskinesia. Since the disorder has only been recognized for two decades and is mostly diagnosed in children, little is known about the disease course. Our purpose was to investigate the disease course of GLUT1DS patients with the classic, complex phenotype from infancy into adulthood. We performed a systematic literature review as well as a cohort study, including GLUT1DS patients aged 18 years and older. The literature search yielded a total of 91 adult GLUT1DS patients, of which 33 patients (one-third) had a complex phenotype. The cohort study included seven GLUT1DS patients with a complex phenotype who were prospectively followed up in our clinic from childhood into adulthood. Our results show that epilepsy is a prominent feature during childhood in classic GLUT1DS patients. During adolescence, however, epilepsy diminishes or even disappears, but new paroxysmal movement disorders, especially paroxysmal exercise-induced dyskinesia, either appear or worsen if already present in childhood. Intellectual disability was not systematically assessed, but cognitive functions appeared to be stabile throughout life. Like children, adolescents may benefit from a ketogenic diet or variants thereof.
Similar content being viewed by others

Introduction
GLUT1 deficiency syndrome (GLUT1DS) is a treatable genetic disorder in which glucose transport over the blood–brain barrier and within the brain is disturbed [1, 2]. GLUT1DS was first recognized in 1991 in two children with seizures, delayed development, and persistently low glucose in the cerebrospinal fluid (CSF) [3]. GLUT1DS can be treated with a ketogenic diet, as ketone bodies serve as alternative fuel for the brain [4].
Over the last 20 years, the knowledge of the phenotypic spectrum of GLUT1DS has increased enormously. The classic and complex phenotype includes intellectual disability, epilepsy, and movement disorders such as spasticity, ataxia, and dystonia [5, 6]. Other, often milder phenotypes are increasingly recognized and these include generalized epilepsies [7, 8], paroxysmal exercise-induced dyskinesia (PED) [9, 10], dystonic tremor [11], and hemolytic anemia [12]. Since the disorder has been recognized for only two decades and is mostly diagnosed in children, little is known about the course of this disease from infancy into adulthood. The vast majority of the adult GLUT1DS patients described in the literature are part of a family with a mild phenotype [9, 13, 14].
To gain more knowledge of the long-term evolution of the classic, complex phenotype of GLUT1DS, we performed a systematic review of the literature and comprehensively studied a cohort that included adult GLUT1DS patients.
Methods
Systematic review
Pubmed was searched (until July 2012) using the MeSH term "glucose transporter type 1" with the subheading "deficiency" and limit "humans", as well as the free search terms "glucose transporter-1 (GLUT-1) deficiency syndrome", "De Vivo disease", and "SLC2A1 gene". Abstracts were screened for articles describing GLUT1DS patients, and if considered relevant, the full article was studied. Relevant papers referred to in the selected articles were also included. We included GLUT1DS patients aged 18 years or older at the time of description in the literature. Patients described more than once were included only once.
Cohort study
The Radboud University Medical Centre in Nijmegen (the Netherlands) is a tertiary referral center with high clinical expertise on GLUT1DS. From the year 2000, all patients diagnosed at or referred to our clinic with GLUT1DS were registered and prospectively followed up in our outpatient clinic.
Patients were included in the present study if the current age was 18 years or older. We only included patients with the classic, complex phenotype of GLUT1DS, defined as intellectual disability in combination with epilepsy or movement disorders. The diagnosis of GLUT1DS was based on the clinical symptoms, in combination with a GLUT1DS CSF profile (i.e., low CSF glucose in combination with a low CSF/blood glucose ratio, and low to normal CSF lactate) [15] and confirmed by SLC2A1 mutation analysis. All patients were invited to our outpatient clinic for the evaluation of current symptoms and signs. Patient history had already been registered in medical records and was complemented by interviewing patients and caregivers. Neurological examination was performed by a (child) neurologist (WGL, MMW).
Results
Systematic review
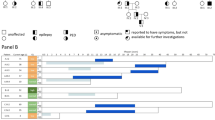
The literature search yielded a total of 46 full articles in English. We studied all articles and found 43 articles describing a total number of 194 GLUT1DS patients, including 91 unique adult patients described in 25 articles. Currently, no official classification of GLUT1DS on clinical grounds exists. We have chosen to classify the patients in three main categories: (1) the classic, complex phenotype of GLUT1DS with intellectual disability in combination with epilepsy or a movement disorder, (2) the ‘epilepsy-dominant’ phenotype, with epilepsy as main symptom, sometimes in combination with a paroxysmal movement disorder such as PED, but without intellectual disability, and (3) the ‘movement disorder-dominant’ phenotype characterized by an isolated movement disorder (mostly PED), without intellectual disability and without epilepsy.
Out of 91 adult GLUT1DS patients described in literature, about one-third (33 patients) had the complex phenotype, one-third (28 patients) had the ‘epilepsy-dominant’ phenotype, and the other third (27 patients) were of the ‘movement disorder-dominant’ phenotype. Furthermore, three asymptomatic adults with affected family members were identified with a SLC2A1 mutation. Out of 33 patients with a complex phenotype, the majority (24 patients) had experienced seizures at some point during their lives. The onset of epilepsy was during childhood in more than half of the patients (19 patients) and during adolescence in three. Ten patients (one-third) still experienced seizures during adulthood. Absence seizures and generalized tonic–clonic seizures were both seen in about one-third (nine and 12 patients, respectively) of the classic GLUT1DS patients during childhood, in contrast to only a small percentage (two and one patient, respectively) during adulthood. Paroxysmal exercise-induced dystonia was found in about two-thirds (21 patients) of the classic GLUT1DS patients, with an onset between 2 and 20 years. Detailed clinical characteristics of all adult GLUT1DS patients identified in the literature are shown in Table 1.
Cohort study
Seven patients with a complex phenotype of GLUT1DS, aged 20–41 years (mean 26.7 years), were included. Patients were prospectively followed at our center for an average of 11 years (range 4–21 years). Three patients were prospectively followed up at our center from childhood (patients 1, 2 and 3), one patient from adolescence (patient 4) and the other three patients from adulthood, with retrospective data being obtained from medical records and caregivers. Patient characteristics are described in Table 2.
Functional outcome
Intellectual disability ranged from mild to moderately severe. Current speech and use of language ranged from normal in three patients to severe dysarthria with only the use of single words in two. Mobility ranged from unassisted walking in three patients to being wheelchair-bound in two. Five patients needed a form of sheltered living with a day care program. Patient 2 (aged 21 years) still lived with her parents and attended regular vocational school. One patient lived independently, and was declared medically unfit for work and worked as a volunteer.
Epilepsy
Six patients experienced epilepsy previously or currently. Seizure onset ranged from the first year of life to childhood. Seizure types included paroxysmal abnormal eye movements, absence epilepsy, focal seizures, atonic attacks, myoclonic seizures, and generalized tonic–clonic seizures. Current seizures were absent or rare in all but patient 1, who still suffered from subtle absences in the morning. Four patients still used one or more anti-epileptic drugs.
Movement disorders
Pyramidal signs were found in all patients, ranging from mild to severe spasticity. Spasticity was noticed at infancy in all but one patient. In patient 4, spasticity was absent until the age of 10 years, with progression to severe spasticity during puberty, after which he became wheelchair-bound for long distances. Progression of spasticity during puberty or adolescence was also seen in three other patients. Ataxia was found in all patients, with an onset during infancy in six patients and during late childhood in one. Progression of ataxia during puberty was seen in three patients. PED was found in all four ambulatory patients, with an onset during adolescence or early adulthood in three. In two patients, PED worsened during adulthood. Subtle, low-frequent PED of the arms during cycling developed in patient 1 at adolescence while on the KD. Paroxysmal oromandibular dystonia was found in patient 5, with an onset at adolescence.
Other paroxysmal events
Other paroxysmal events, all in individual cases, included: episodes with hemiplegic attacks and loss of consciousness during childhood; autonomic attacks with sweating and yawning, combined with generalized weakness and impaired consciousness with an onset during childhood and progression into adolescence; paroxysmal dysarthria during episodes of PED with an onset during childhood and stabile frequency into adulthood; action myoclonus with an onset during adulthood and progression during the first years after onset; and painful cramps in the legs during the night with an onset during adulthood.
Treatment with a ketogenic or modified Atkins diet
The ketogenic diet was initiated in patient 1 at the age of 8 years, with a 90 % reduction of daily absence seizures. In patient 2, the ketogenic diet was initiated at the age of 9 years, after which daily absence seizures totally disappeared. The diet was stopped due to incompliance at the age of 13 years, after which absence seizures initially re-occurred and spontaneously disappeared during adolescence. The modified Atkins diet was initiated in this patient at the age of 20 years because of the progression of PED, after which PED totally disappeared as long as this patient was compliant with the diet [16]. In patient 3, the ketogenic diet was started during puberty with a >90 % reduction of myoclonic and complex partial seizures. The diet was stopped after a year because of non-compliance. At the age of 23 years, the modified Atkins diet was introduced because of PED. Despite good compliance and ketosis, the diet did not have a positive effect on the PED and was stopped after 3 months. Treatment with a low dose of oxcarbazepine was started with total resolution of PED. A modified Atkins diet was started in patients 4 and 5 during adulthood, after which their respective symptoms of daily complex partial seizures and paroxysmal oromandibular dystonia completely resolved [16]. A modified Atkins diet was started in patient 6 as well, at the age of 27 years, with a reduction of action myoclonus and improvement of alertness, but was stopped after 3 months according to the patient's wishes. Patient 7 was never on a ketogenic or modified Atkins diet, but preferred a regular diet with frequent intake of carbohydrate-containing snacks such as dextro energy sweets. He, however, gained a lot of weight and is now trying to lose weight with the help of a dietitian.
Discussion
We studied the disease course from infancy into adulthood in patients with the classic, complex phenotype of GLUT1DS. Whereas epilepsy is a prominent feature during childhood, seizures typically decrease during adolescence, irrespective of treatment with anti-epileptic drugs or a ketogenic diet. On the other hand, movement disorders may worsen or appear during adolescence, especially paroxysmal movement disorders such as PED, but the progression of spasticity and ataxia may also be observed.
The functional outcome for patients with the classic phenotype of GLUT1DS is very variable. Although many patients become ambulatory, a substantial number of patients require the use of a wheelchair. Furthermore, whereas most patients need some form of sheltered living, independent living after attending regular school is possible for some.
Early introduction of a ketogenic diet and maintenance into adulthood is recommended in patients with GLUT1DS to meet the energy demands of the developing brain [17, 18]. An early start of the ketogenic diet is thought to have a positive influence on the final outcome, but this is considered difficult to prove scientifically. Our study demonstrates a favorable natural course of the epilepsy in GLUT1DS and indicates that withdrawal of anti-epileptic drugs and the ketogenic diet could be considered after adolescence. Four of our patients remained on anti-epileptic drugs during adulthood, but three of those patients are currently on a withdrawal scheme. Patient 3 preferred to continue treatment with anti-epileptic drugs, despite the total absence of seizures over the last 5 years.
On the other hand, our cohort study shows that worsening or onset of paroxysmal movement disorders during adolescence or adulthood can be reason to continue or (re)-introduce a ketogenic diet, with the modified Atkins diet as a good and feasible alternative [16]. In some patients with relatively mild symptoms, a regular diet with frequent carbohydrate-containing snacks can be effective. However, such a diet easily leads to obesity (as was the case in patient 7), and therefore our advice is to discuss the possibility of a modified Atkins diet in all adult GLUT1DS patients.
The classification of patients with GLUT1DS into subcategories remains difficult, and several subclassification systems have already been proposed [17, 19–21]. Although GLUT1DS represents a spectrum of phenotypes with large overlap between different groups, our systematic review shows that a subclassification based on the main (and often presenting) symptom does have practical utility. Patients with the ‘complex’, ‘epilepsy-dominant’, or ‘movement-disorder dominant’ phenotype subcategory have different disease courses, which is important information for further counseling. Interestingly, we did not encounter GLUT1DS patients with isolated intellectual disability (i.e., without epilepsy or movement disorders). Hypothetically, such patients are not recognized and thereby missed as GLUT1DS patients. More likely, however, is that SLC2A1 mutations do not present with pure intellectual disability, but are always accompanied by epilepsy or movement disorders. In exome sequencing studies for de novo mutations in patients with isolated intellectual disability, SLC2A1 mutations have not been described, suggesting that this is not a major gene involved in isolated intellectual disability.
Only 36 % of the adult patients described in the literature had a complex phenotype. Most likely, this relatively small number is biased by the fact that in recent years, only patients with a novel phenotype, such as an isolated movement disorder, were reported. Also, the classic, complex phenotype generally occurs de novo, while milder phenotypes are transmitted in an autosomal dominant manner.
A possible bias of a prospective cohort study is that patients with a favorable outcome tend to discontinue medical follow-up and are thereby missed. In our cohort, however, all GLUT1DS patients diagnosed or referred to our hospital have been systematically registered and were prospectively followed up over the last 12 years. No patients withdrew from the study during the follow-up.
Our results illustrate that in patients with a complex phenotype of GLUT1DS, the dominant symptom may change with age, with epilepsy typically found to be the most disabling symptom during childhood, and onset or worsening of (paroxysmal) movement disorders during adolescence and early adulthood. This might be related to physiological changes in local cerebral glucose utilization during the development of the brain. Position emission tomography (PET) studies have shown that the pattern of cerebral glucose utilization undergoes large changes from birth to adulthood [22]. During the neonatal period, there is a relatively low cerebral metabolic rate of glucose utilization over most of the cerebral cortex. Between birth and the age of 4 years, a dramatic increase in cortical glucose utilization is seen, twice the rate in adults. From the age of 4–10 years, glucose consumption rates remain very high, followed by a gradual decline to adult values by the age of 16–18 years. Interestingly, the changing pattern of glucose utilization during development parallels the occurrence and disappearance of epilepsy during childhood and adolescence: the onset of seizures is generally during infancy (rarely neonatal), with a peak during childhood and a decrease or even remission during adolescence and adulthood.
A possible relation between the age at onset of paroxysmal movement disorders such as PED in GLUT1DS and changes in local cerebral glucose utilization is more complex. First of all, it is not totally clear which part of the brain is responsible for the occurrence of PED. Cerebral 18F-fluorodeoxyglucose (FDG) PET studies in GLUT1DS during an episode of PED suggest that a disordered glucose metabolism in the corticostriate pathway might play a role [9, 23]. However, cerebral perfusion studies using single-photon emission computed tomography (SPECT) in patients with PED that is not necessarily due to GLUT1DS have suggested frontal lobe and cerebellar involvement in the pathophysiology of PED [24]. The possible role of the cerebellum during exercise is further supported by the fact that studies in rats have shown that during exercise, a relatively larger increase in glucose metabolism is seen in the cerebellum, compared to the motor cortex and basal ganglia [25]. This suggest that the cerebellum might be especially vulnerable for a glucose deficit during exercise. Over the years, it has become clear that a cerebellar defect can singlehandedly lead to dystonia [26]. The exact neuro-anatomical substrate of PED in GLUT1DS is thus not completely clear, and further investigation by means of neuroimaging techniques such as magnetic resonance spectroscopy are essential to gain more insight.
With the increasing number of adult GLUT1DS patients currently being reported in the literature and systematically followed up in tertiary referral centers like ours, the long-term disease course of GLUT1DS is gradually becoming clearer. Our data facilitate the counseling of patients with GLUT1DS and their caregivers. In contrast to many other neurometabolic disorders, GLUT1DS is not a progressive neurodegenerative disorder. Interestingly, some brain functions appear stabile (e.g., cognition and behavior), while others diminish over time (like epilepsy) or appear only with increasing age (like PED). Fortunately, like children, adolescents and adults may also benefit from treatment with the ketogenic or modified Atkins diet [16].
References
Pascual JM, Wang D, Lecumberri B et al (2004) GLUT1 deficiency and other glucose transporter diseases. Eur J Endocrinol 150:627–633
Wang D, Pascual JM, Yang H et al (2006) A mouse model for Glut-1 haploinsufficiency. Hum Mol Genet 15:1169–1179
De Vivo DC, Trifiletti RR, Jacobson RI et al (1991) Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 325:703–709
Klepper J (2008) Glucose transporter deficiency syndrome (GLUT1DS) and the ketogenic diet. Epilepsia 49(Suppl 8):46–49
Pearson TS, Akman C, Hinton VJ, Engelstad K, De Vivo DC (2013) Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr Neurol Neurosci Rep 13:342
Tzadok M, Nissenkorn A, Porper K et al (2013) The many faces of Glut1 deficiency syndrome. J Child Neurol (Epub ahead of print)
Arsov T, Mullen SA, Damiano JA et al (2012) Early onset absence epilepsy: 1 in 10 cases is caused by GLUT1 deficiency. Epilepsia 53:e204–e207
Arsov T, Mullen SA, Rogers S et al (2012) Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol 72:807–815
Suls A, Dedeken P, Goffin K et al (2008) Paroxysmal exercise-induced dyskinesia and epilepsy is due to mutations in SLC2A1, encoding the glucose transporter GLUT1. Brain 131:1831–1844
Afawi Z, Suls A, Ekstein D et al (2010) Mild adolescent/adult onset epilepsy and paroxysmal exercise-induced dyskinesia due to GLUT1 deficiency. Epilepsia 51:2466–2469
Roubergue A, Apartis E, Mesnage V et al (2011) Dystonic tremor caused by mutation of the glucose transporter gene GLUT1. J Inherit Metab Dis 34:483–488
Flatt JF, Guizouarn H, Burton NM et al (2011) Stomatin-deficient cryohydrocytosis results from mutations in SLC2A1: a novel form of GLUT1 deficiency syndrome. Blood 118:5267–5277
Weber YG, Kamm C, Suls A et al (2011) Paroxysmal choreoathetosis/spasticity (DYT9) is caused by a GLUT1 defect. Neurology 77:959–964
Mullen SA, Suls A, De Jonghe P, Berkovic SF, Scheffer IE (2010) Absence epilepsies with widely variable onset are a key feature of familial GLUT1 deficiency. Neurology 75:432–440
Leen WG, Wevers RA, Kamsteeg EJ et al (2013) Cerebrospinal fluid analysis in the work-up of GLUT1 deficiency syndrome. JAMA Neurol. doi: 10.1001/jamaneurol.2013.3090 (Epub ahead of print)
Leen WG, Mewasingh L, Verbeek MM et al (2013) Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov Disord 10:1439–1442
Ramm-Pettersen A, Nakken KO, Skogseid IM et al (2013) Good outcome in patients with early dietary treatment of GLUT-1 deficiency syndrome: results from a retrospective Norwegian study. Dev Med Child Neurol 55:440–447
Klepper J (2011) GLUT1 deficiency syndrome in clinical practice. Epilepsy Res 100:272–277
Leen WG, Klepper J, Verbeek MM et al (2010) Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 133:655–670
Wang D, Pascual JM, Yang H et al (2005) Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol 57:111–118
Brockmann K (2009) The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev 31:545–552
Chugani HT (1998) A critical period of brain development: studies of cerebral glucose utilization with PET. Prev Med 27:184–188
Pascual JM, van Heertum RL, Wang D, Engelstad K, De Vivo DC (2002) Imaging the metabolic footprint of Glut1 deficiency on the brain. Ann Neurol 52:458–464
Kluge A, Kettner B, Zschenderlein R et al (1998) Changes in perfusion pattern using ECD-SPECT indicate frontal lobe and cerebellar involvement in exercise-induced paroxysmal dystonia. Mov Disord 13:125–134
Vissing J, Andersen M, Diemer NH (1996) Exercise-induced changes in local cerebral glucose utilization in the rat. J Cereb Blood Flow Metab 16:729–736
Sadnicka A, Hoffland BS, Bhatia KP, van de Warrenburg BP, Edwards MJ (2012) The cerebellum in dystonia—help or hindrance? Clin Neurophysiol 123:65–70
Acknowledgments
W.G. Leen was supported by the Netherlands Organization for Scientific Research (NWO; project number 92003529; http://www.nwo.nl).
Conflicts of interest
On behalf of all authors, the corresponding author states that there are no conflicts of interest.
Ethical standard
This study has been approved by the appropriate ethics committee and has therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Leen, W.G., Taher, M., Verbeek, M.M. et al. GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol 261, 589–599 (2014). https://doi.org/10.1007/s00415-014-7240-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-014-7240-z