
Abstract
We have investigated the extent and pattern of immunostaining for ubiquitin protein (UBQ) in 60 patients with frontotemporal lobar degeneration (FTLD) with ubiquitin-positive, tau-negative inclusions (FTLD-U), 37 of whom were ascertained in Manchester UK and 23 in Newcastle-Upon-Tyne, UK. There were three distinct histological patterns according to the form and distribution of the UBQ pathology. Histological type 1 was present in 19 patients (32%) and characterised by the presence of a moderate number, or numerous, UBQ immunoreactive neurites and intraneuronal cytoplasmic inclusions within layer II of the frontal and temporal cerebral cortex, and cytoplasmic inclusions within granule cells of the dentate gyrus; neuronal intranuclear inclusions (NII) of a “cat’s eye” or “lentiform” appearance were present in 17 of these patients. In histological type 2 (16 patients, 27%), UBQ neurites were predominantly, or exclusively, present with few intraneuronal cytoplasmic inclusions within layer II of the cerebral cortex, while in histological type 3 (25 patients, 42%), UBQ intraneuronal cytoplasmic inclusions either within the cortical layer II or in the granule cells of the dentate gyrus, with few or no UBQ neurites, were seen. In neither of these latter two groups were NII present. The influence of histological type on clinical phenotype was highly significant with type 1 histology being associated clinically with cases of frontotemporal dementia (FTD) or progressive non-fluent aphasia (PNFA), type 2 histology with semantic dementia (SD), and type 3 histology with FTD, or FTD and motor neurone disease (MND).
Similar content being viewed by others


Introduction
Frontotemporal lobar degeneration (FTLD) is a descriptive term that refers to a clinically and pathologically heterogeneous group of non-Alzheimer forms of dementia with onset of illness usually before 65 years of age [21, 29]. While frontotemporal dementia (FTD) is the main clinical syndrome arising from the degeneration of the frontal and temporal lobes in FTLD, related disorders of semantic dementia (SD), progressive non-fluent aphasia (PNFA) stem from differing (to FTD) topographical distributions of similar underlying pathologies [21, 29]. When the frontal and temporal lobes are bilaterally affected, the syndrome of FTD emerges, while in SD the temporal lobes are bilaterally affected and in PNFA the left cerebral hemisphere is preferentially affected [21, 29]. When the behavioural and personality changes of FTD are accompanied by clinical Motor Neurone Disease, the syndrome of frontotemporal dementia with Motor Neurone Disease (MND) (FTD + MND) emerges [21, 29].
There are also pathological heterogeneities within FTLD. About 45% cases contain insoluble tau proteins in their brains in the form of intraneuronal neurofibrillary tangles or Pick bodies [28, 30]: many such cases are associated with mutations in the tau (MAPT) gene (see [19] for review). However, numerous pathological investigations on large series of patients with FTLD [2, 6, 10–15, 17, 20, 28, 30] indicate that a tau-negative histology is a frequent, if not the most common, change underlying FTLD, with the great majority of cases showing ubiquitin (UBQ)-positive pathology in the form of intraneuronal inclusions and/or neuritic changes in cerebral cortex and the hippocampus, a pathology now referred to as FTLD-U or FTLD-MND when clinical MND is also present. In some FTLD-U cases, neuronal intranuclear inclusions of a “cat’s eye” or “lentiform” appearance have been described [16, 32], especially in those cases with autosomal dominant inheritance of disease and linkage to chromosome 17 [8, 18, 24, 25].
In this study, we have investigated the extent and pattern of UBQ immunostaining in 60 patients with FTLD, 37 of whom were ascertained in Manchester UK, and 23 in Newcastle-Upon-Tyne, UK. We have found three distinct patterns of UBQ staining, and have correlated these with clinical phenotype and MAPT haplotype.
Materials and methods
Thirty seven cases with FTLD-U or FTLD-MND were selected for this present study from a series of 73 consecutive patients dying since 1987 within North West region of United Kingdom, fulfilling clinical diagnostic criteria for sporadic and familial FTLD [4, 23] (patients #1–37, Table 1). We have recently described the histopathological characteristics of these patients [17, 28]. Of these 37 patients, 25 (68%) were male, 12 (32%) were female. Mean age at onset of disease was 57.7 ± 7.5 years, and mean duration of illness was 8.1 ± 4.2 years. A further 23 cases of pathologically diagnosed FTLD-U were ascertained through the Brain Retrieval Programme at IAH, Newcastle General Hospital (patients #38–60, Table 1). Retrospectively, the clinical diagnosis in these cases was compatible with sporadic and/or familial FTLD [4, 23]. Of these 23 patients, 13 (57%) were male, 10 (43%) were female. Mean age at onset of disease was 59.7 ± 17.8 years, and mean duration of illness was 5.2 ± 2.9 years. The Manchester patients did not differ from the Newcastle patients in terms of gender distribution (χ2 = 0.75, P = 0.39) or age at onset (t = 0.59, P = 0.56). However, they had a longer duration of illness (t = 2.79, P = 0.007). At both Centres, all brains had been collected in accordance with Local Ethical Committee approval.
Brains had been fixed in 10% buffered formalin for a period of between 1–3 months after which blocks of tissue were cut from frontal (Brodmann areas 8/9), and (except in patient #38) temporal (Brodmann areas 21/22) cortex and hippocampus (at the level of the geniculate bodies), and processed routinely into paraffin wax. Sections were cut at a thickness of 5 μm. Although UBQ pathology is also widely present within the striatum in cases of FTLD-U [13], this region was not always available, and therefore not included in the present study. Sections of frontal cortex from both Manchester and Newcastle series, and those of temporal cortex and hippocampus from the Manchester series alone, were immunostained for UBQ using an automated staining procedure [17], using a polyclonal anti-UBQ antibody (Dako, Glostrup, Denmark, 1:500). Sections of temporal cortex and hippocampus from the Newcastle series were immunostained in Newcastle, again using the polyclonal DAKO UBQ antibody, at the same dilution (1:500).
The UBQ immunostained sections were viewed by experienced Neuropathologists in Manchester (DMAM), Newcastle (AB) and Vancouver (IRAM) and were classified into UBQ histological type by consensus according to the form and distribution of UBQ pathological changes. The severity of UBQ pathology within each histological type, both as the extent of neuritic changes or neuronal cytoplasmic inclusions within layer II of the frontal and the temporal cortex, or neuronal cytoplasmic inclusions within granule cells of the dentate gyrus of the hippocampus was rated semiquantitatively according to (0 = absent; 1 = rare; 2 = mild; 3 = moderate; 4 = severe). Rating was performed independently by two observers (DMAM and IRAM), with the UBQ score being averaged where any disagreement arose. Scores for the extent of neuritic changes and neuronal cytoplasmic inclusions in frontal and temporal cortex, and neuronal cytoplasmic inclusions in hippocampus, were then summed to give an overall cerebral cortical UBQ severity rating (maximum = 20).
All statistical tests were performed using SSPS 13.0.
Results
Histological phenotypes
On the basis of appearance in UBQ immunostained sections, three histological phenotypes were discerned:
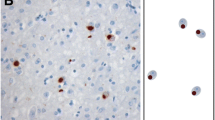
Type 1: This was present in 19 patients (32%) (patients #1–14 and #38–42) . In these, there was a moderate number, or numerous, UBQ immunoreactive neurites and intraneuronal cytoplasmic inclusions within layer II of the cerebral cortex (Fig. 1a, Table 1). Cytoplasmic inclusions within granule cells of the dentate gyrus were variable, ranging from a few to many (Fig. 1b, Table 1). Neuronal intranuclear inclusions (NII) of a “cat’s eye” or “lentiform” appearance [16, 32] were present in 17 of these patients (Fig. 1c, Table 1). These were never very numerous in any patient, ranging from about ten per section (of frontal or temporal cortex) in some patients down to only a single NII in others. Only in a single patient (patient #2) was a single NII seen within dentate gyrus granule cells (Fig. 1d), and in another patient (patient #3) NII were seen within two pyramidal cells in area CA4 of hippocampus (one shown in Fig. 1e).

Type 1 histology: In patient #1 there are numerous, UBQ immunoreactive neurites and intraneuronal cytoplasmic inclusions within layer II of the cerebral cortex (a) though UBQ cytoplasmic inclusions within granule cells of the dentate gyrus were relatively few (b). Neuronal intranuclear inclusions (NII) of a “cat’s eye” or “lentiform” appearance were also seen in pyramidal cells of layer II of cerebral cortex in this patient (c). Only in a single patient (patient #2) was a single NII seen within hippocampus dentate gyrus granule cells (d), and in another patient (patient #3) NII were seen within pyramidal cells in area CA4/5 of hippocampus (e). Immunoperoxidase-haematoxylin, ×20 (a), ×40 (b–e) microscope magnification
Type 2: This was present in 16 patients (27%) (patients #15–23 and #43–49). In these, UBQ neurites were predominantly, or exclusively, present within layer II of the cerebral cortex with few or no intraneuronal cytoplasmic inclusions (Fig. 2a, Table 1). Cytoplasmic inclusions within granule cells of the dentate gyrus were variable, being few or absent in eight patients, moderate in three patients, but numerous in four others (Fig. 2b, Table 1); the hippocampus was not available for investigation in the other case. NII were not present in any of these 16 patients.

Type 2 histology: In patients #22 and #19, there are numerous UBQ neurites predominantly, or exclusively, present within layer II of the cerebral cortex with few or no intraneuronal cytoplasmic inclusions (a and c, respectively). UBQ cytoplasmic inclusions within granule cells of the dentate gyrus were also numerous in patient #22 (b), but were less common in patient #19 (d) Immunoperoxidase-haematoxylin, ×20 (a, c), ×40 (b, d) microscope magnification
Type 3: In the remaining 25 patients (42%) (patients #24–37 and #50–60), UBQ neuronal cytoplasmic inclusions were predominantly, or exclusively, present within the brain with only relatively few UBQ neurites being present. Two subtypes were identified. In 12 patients, with type 3a histology (patients #24–29 and #50–55), UBQ inclusions were numerous within neurones of layer II of the frontal and temporal cortex (Fig. 3a, Table 1), but only in 6 of these patients were cytoplasmic inclusions within granule cells of the dentate gyrus numerous (Fig. 3b, Table 1). In the other 13 patients, with type 3b histology (patients #30–37 and #56–60), UBQ cytoplasmic inclusions within granule cells of the dentate gyrus was the major pathology (Fig. 3c, Table 1), with relatively few or no neuronal cytoplasmic inclusions (and neurites) being present within the cerebral cortex (Fig. 3d). Again, NII were not present in any patient.

Type 3 histology: In patient #28 with type 3a histology, UBQ neuronal cytoplasmic inclusions are predominantly, or exclusively, present within the brain with only relatively few UBQ neurites being present (a). UBQ inclusions were also sometimes numerous within granule cells of the dentate gyrus numerous (b). In patient #32 there were few neuronal UBQ cytoplasmic inclusions and neurites within the cerebral cortex (c), though UBQ cytoplasmic inclusions were widespread within granule cells of the hippocampus dentate gyrus (d). Immunoperoxidase-haematoxylin, ×20 (a, c), ×40 (b, d) microscope magnification
Patients with type 2 histology were clearly differentiated from those with type 1 histology according to the relative balance between UBQ neuronal cytoplasmic inclusions and UBQ neurites within the cerebral cortex. The presence of NII per se did not act as a sine qua non for type 1 histology (two patients with type 1 histology did not show NII), nor did the number or presence of hippocampal cytoplasmic inclusions influence such differentiation. In type 1 histology, UBQ neuronal cytoplasmic inclusions and neurites were both numerous within layer II of the cerebral cortex, whereas in type 2 histology the overwhelming preponderance of UBQ neuritic pathology relative to UBQ neuronal inclusions was the deciding factor. The presence of numerous UBQ neuronal inclusions either within the cerebral cortex or the hippocampus (or both) in the relative or total absence of a UBQ neuritic pathology marked type 3 histology from that of either type 1 or type 2 histology.
The mean total UBQ score was significantly greater (F 2,56 = 12.3, P < 0.001) in cases with type 1 histology (12.5 ± 4.2) than for cases with type 2 (7.6 ± 3.4) or type 3 ( i.e. with 3a and 3b type histologies combined) (6.6 ± 4.2), which did not differ from each other.
Demographics
Of the 19 patients with type 1 UBQ histology, 11 (58%) were male and 8 (42%) were female. Mean age at onset of disease was 62.0 ± 7.7 years and mean duration of disease was 8.4 ± 4.1 years. Of the 16 patients with type 2 UBQ histology, 8 (50%) were male and 8 (50%) were female. Mean age at onset of disease was 60.1 ± 13.5 years and mean duration of disease was 8.1 ± 4.0 years in those 14/16 patients where this information was available. Of the 25 patients with type 3 UBQ histology, 19 (76%) were male and 6 (24%) were female. Mean age at onset of disease was 54.8 ± 13.5 years and mean duration of disease was 5.4 ± 3.5 years.
The preponderance of males compared to females associated with type 3 histology was statistically significant (binomial test, P = 0.015). Male to female ratios did not differ significantly for other histological types. Numerical differences in age at onset of cases with each of the three histological phenotypes did not reach statistical significance (F 2,55 = 2.16, P = 0.13). However, there was a significant effect of histological type on duration of illness (F 2,55 = 3.83, P = 0.03). Post-hoc Bonferroni tests showed that cases with type 3 histology had a significantly shorter duration than cases with type 1 histology (P = 0.04).
Relationship to clinical phenotype
Although, retrospectively the Newcastle cases had a clinical diagnosis compatible with FTLD, these cases had not been prospectively assessed with the primary purpose of characterising the neuropsychological deficit. It was therefore only possible to examine relationships with clinical subtype within the Manchester series of 37 cases. Eight of 14 patients with type 1 histology had clinical FTD, 5 had PNFA and 1 SD. One of nine patients with type 2 histology had FTD, one had PNFA but seven had SD. Seven of 14 patients with type 3 histology had FTD, 6 had FTD + MND and 1 had PNFA. Putting it another way, eight of eleven patients with FTD had type 1 histology and three had type 3 histology. Five of seven patients with PNFA had type 1 histology and one each had types 2 and 3 histology. Seven of eight patients with SD had type 2 histology and one had type 1 histology. All 6 patients with FTD + MND had type 3 histology. Hence, type 1 pathology was mostly associated with cases of FTD or PNFA, whereas type 2 histology tended to be associated mostly with cases of SD, and type 3 pathology with cases of FTD or FTD + MND. The influence of histological type on clinical phenotype was highly significant (χ2 = 33.7, P < 0.0001)
Relationship to genotype
MAPT haplotype and genotype data was available for 32/37 Manchester patients and 17/23 Newcastle patients. MAPT haplotype frequency did not differ across any of the three histological groups (Table 2). There was no overall influence of histological type on genotype frequency (χ2 = 3.98, P = 0.14). However, the comparison of histological types 2 and 3 alone reached statistical significance (χ2 = 3.83, P = 0.05), suggesting that H1H1 genotype was more likely to be associated with type 2 histology and H1H2 with type 3 histology. More importantly, however, when combining all three histological groups, total UBQ score tended to increase (F 2,46 = 56.7, P = 0.047) with possession of MAPT H2 haplotype in a dose-dependent way (H1H1 = 8.4 ± 3.5, H1H2 = 9.6 ± 4.9, H2H2 = 16.0 ± 4.2).
Discussion
In recent times there have been numerous investigations [2, 6, 10, 17, 20, 28, 30] indicating that a tau-negative, ubiquitin-positive pathology, known as FTLD-U, is a frequent, if not the most common, histological change underlying FTLD. Nonetheless, in most studies [2, 6, 10–12, 14, 15, 17, 20, 28, 30], little or no attempt has been made to differentiate cases according to the phenotypic appearance of the UBQ changes, with all cases being simply referred to as FTLD-U. Only in one previous study [13] did the authors correlate the appearance of the ubiquitin pathology with clinical phenotype, but then mainly from the perspective of differentiating between cases of FTD with or without accompanying MND. In this latter study, this was done according to differences in the density and morphology of UBQ changes in the cerebral cortex, hippocampus and striatum [13]. In the present study, we have classified 60 cases of FTLD-U drawn from two UK Centres (Manchester and Newcastle) into three distinct histological subtypes according to the appearance and anatomical distribution of the UBQ pathology within the cerebral cortex and hippocampus. We have been able to extend previous observations [13] and show that clinical representations of FTLD other than FTD + MND (i.e. FTD, SD and PNFA) are strongly associated with each of the different histological subtypes. The results of our study may have important nosological implications for the classification of FTLD.
The histological subtype we have designated as type 1 is characterised by many UBQ immunoreactive neurites, and UBQ intraneuronal cytoplasmic inclusions with “cat’s eye” or “lentiform” NII, within layer II of the cerebral cortex, and variable numbers of UBQ cytoplasmic inclusions within granule cells of the dentate gyrus. Nineteen patients (32%) showed type 1 histology, 17 with NII. This type of histology was typically seen in patients with clinical FTD or PNFA. On the basis of their examinations of UBQ immunostained sections of cerebral cortex, hippocampus and striatum, Katsuse and Dickson [13] described one form of UBQ pathology, which they termed FTLD-MNI (FTLD with motor-neuron disease (MND)-type inclusions but without MND) in 47/52 patients with FTLD-U and another form which they called FTLD-MND (FTLD with MND). Although here we did not examine the striatum, their descriptions of FTLD-MNI and FTLD-MND [13] bear close resemblance to those which we have termed type 1 and type 3 histology, respectively. Nonetheless, in contrast to present findings, where NII were universal in type 1 cases, these were reported by Katsuse and Dickson to occur in only 26 of their FTLD-MNI cases [13]. It is possible that selection criteria account for these differences. In the present study, we differentiated cases of type 1 histology from those with type 2 histology (see above for criteria) in whom NII were not seen, whereas in the study by Katsuse and Dickson [13], because they did not attempt to further subtype their FTLD-MNI cases, it is possible that these could have included cases of (our) type 2 histology in whom NII would not have been anticipated.
In the present study, type 2 histology, with abundant UBQ neurites but few or no cytoplasmic inclusions or NII, was the most common histology present in patients with SD-8/9 Manchester patients with SD showed type 2 histology and 8/9 patients with type 2 histology had SD. FTLD-U had been previously reported by Rossor et al [26] to be the underlying histology in three patients with SD, and from the histological descriptions provided it would appear that these three patients also displayed UBQ pathological changes similar to type 2 histology described here, although this was not so remarked upon at the time.
Type 3 histology, characterised by numerous neuronal UBQ cytoplasmic inclusions but few or no UBQ neurites was seen in all six Manchester patients with FTD + MND. Although others [3, 13] have suggested that the type 1 histology may be present in some cases of FTD + MND, in the present series such cases were not encountered. However, it is accepted that the small numbers of such patients studied here may have precluded the finding of cases with alternative UBQ histologies. In four of these (patients# 25–28) both cerebral cortical and hippocampal neurones were extensively involved, though in the other two (patients #36 and 37) only hippocampal neurones were severely affected. However, there were eight other patients with this type of histology, albeit in most instances restricted to hippocampus, none of whom showed any clinical or pathological evidence of MND. Seven of these patients had FTD alone and one had PNFA, emphasising the overlapping pathology between FTD and FTD + MND, particularly as far as the hippocampus is concerned [31].
When we compared duration of illness across the histological phenotypes we found that it was shorter in patients with type 3 histology than in other patients. These findings are most likely explained by the inclusion of those patients with FTD + MND within this histological subtype, for whom a shorter duration of illness has been previously reported [6, 10, 12, 22]. In such individuals it is likely that the disease is terminated early through the presence of bulbar muscle wasting and respiratory failure consequent upon MND.
What we have presently termed type 1 histology occurs in certain families with autosomal dominant transmission of disease known to be linked to chromosome 17q21 [8, 18, 24, 25]. Recent work [1, 6] has shown that FTLD in many of these latter families is caused by mutations in progranulin gene (PGRN), located just 1.8 mb distant from MAPT locus on chromosome 17q21. Such observations raise the possibility that type 1 histology, and perhaps the NII component thereof, might act as a surrogate marker of PGRN mutation-related FTLD. This argument is supported by several observations.
Firstly, that two of the present patients with FTLD reported in Baker et al. [1], one with Q130SfsX124 mutation and one with Q468Xmutation (patients #4 and #12, respectively in the present study), in whom the disease was not previously known to be linked to chromosome 17, did indeed have this pathology. Since then we have further found in patient #13 the C31LfsX34 mutation reported initially in a Canadian family UBC-17 with chromosome 17-linked tau negative histology and published in Baker et al. [1], and a novel PGRN mutation (V452WfsX38) in patient #7 (Pickering-Brown, unpublished data), both with type 1 histology. We are continuing to analyse the remaining ten patients with this histological subtype for PGRN mutations to provide further support for this viewpoint. Furthermore, since the original studies by Baker et al. [1] and Cruts et al. [6], many other additional FTLD-U cases with PGRN mutations have also been identified by other workers, and NII were present in all cases where post mortem brain analysis had been done (Mackenzie–manuscript submitted).
Secondly, none of the 16 patients with type 2 histology showed NII, even though there was positive family history of FTLD in two of these (patients #43 and #47). So far, we have not detected PGRN mutation in any of these 16 patients, nor have we found any such mutations in a further 25 clinically confirmed living patients with SD (most of whom can be presumed to have type 2 histology) (Pickering-Brown, unpublished data), suggesting PGRN mutations are unlikely to underpin this form of UBQ pathology. Nonetheless, paradoxically, patient #14 with SD did in fact show type 1 histology and had a positive family history; PGRN analysis in this patient has not as yet revealed PGRN mutation (Pickering-Brown, unpublished data).
Thirdly, NII were not present in any patient with type 3 histology, including the six patients with FTD + MND. Similarly, although Katsuse and Dickson [13] reported NII to be present in 26/43 cases of FTLD-U, none of their cases had clinical and pathological MND. However, in another study of 34 patients with FTLD-U, 11 patients showed NII of whom 3 had FTD and clinical and pathological MND [3]. It is not known in either of these latter studies whether any or all such cases with NII bore PGRN mutation. We have so far been unable to detect PGRN mutation in any of the present 6 clinically and pathologically confirmed cases of FTD + MND, nor have we found any such mutations in a further 22 clinically confirmed living patients with FTD + MND and presumed type 3 histology (Pickering-Brown, unpublished data).
However, the finding of ubiquitinated NII in patients with Inclusion Body Myopathy with Pagets Disease of bone and FTD (IBMPFD) associated with mutations in the valosin-containing protein gene (VCP) [7, 26] apparently similar to those seen in FTLD would argue against specificity of UBQ NII in FTLD as markers of PGRN mutations. However, although NII in VCP mutations contain VCP protein [7, 26] and may superficially resemble those in FTLD cases with PGRN mutation, the latter do not contain VCP protein [28]. Moreover, IBMPFD is an extremely rare disorder, and can be readily distinguished from FTLD both clinically and pathologically, in terms of the nature and distribution of UBQ pathology [7]. Hence, patients with “cat’s eye” NII, within the context of clinical FTD not associated with muscle or bone abnormality, are unlikely to possess VCP mutation.
Furthermore, NII in FTLD are not always observed in the context of a previous positive family history. Although in the present study linkage analysis had not been performed, 15/17 patients with NII had shown positive family history consistent with autosomal dominance. Nonetheless, two patients showed no previous family history, and the presence of NII in apparently sporadic cases has been noted by others [3, 13]. On face value, such observations would again argue against NII being pathognomic for PGRN mutations, though in at least some cases included in the latter studies [3, 13] PGRN mutations have in fact now been found (Mackenzie, personal communication). Hence, whether there are indeed sporadic FTLD-U cases with NII but without PGRN mutations must remain an open question until all such potential cases have undergone genetic analysis and the absence of such a mutation definitely ruled out.
Hence, current data would strongly suggest that the presence of NII in FTLD is indicative of PGRN mutation, though this conclusion must remain tentative until all cases with these pathological structures, both within this present study and in those of other research groups, have been subjected to genetic analysis and the presence or absence of mutation defined. Nonetheless, should this relationship between NII and PGRN mutations turn out to be correct, then present data showing type 1 histology to be present in 17/60 patients with FTLD-U in the Manchester and Newcastle combined series (present in 14/37 Manchester patients with FTLD-U, and in 14/73 Manchester patients with FTLD per se) would suggest that PGRN mutations are a more common cause of (inherited) FTLD than those with MAPT mutations, outnumbering the latter by a factor of 2:1 (only 8/73 Manchester patients with FTLD had MAPT mutations [17, 28]).
Interestingly, a number of FTLD clinical phenotypes were embraced by type 1 histology, but principally FTD and PNFA. Indeed, patients #4 and #7 with Q468X and V452WfsX38 mutation, respectively, displayed FTD phenotype, whereas patients #12 and #13 with Q130Sfx124 and C31LfsX34 mutations showed PNFA. Such observations argue that although type 1 histology might be associated per se with PGRN mutations (vide supra) the brain topographic distribution of that form of UBQ pathology may depend upon factors other than the actual mutation, since it has been proposed that all PGRN mutations induce the same pathophysiological haploinsufficiency through the creation of a null allele [1, 6].
Finally, although none of the cases studied here were associated with MAPT mutations, there appeared to be variations in frequency of MAPT haplotypes and genotypes between the different histological subtypes. For example, type 2 histology was associated with a significantly higher H1H1 frequency (but not H1 haplotype frequency), and type 3 histology with H1H2 genotype. By contrast, the extent of UBQ pathology (irrespective of histological subtype) increased with possession of H2 allele. Such observations suggest MAPT haplotype might play a part in modulation of the overall degree of UBQ pathology and/or the histological form manifested. However, these observations are based on small sample sizes, especially those relating to H2 allele, and will require validation in further, and larger, cohorts before they can be considered to be substantive.
What the ubiquitinated protein present within the neuronal cytoplasmic inclusions, NII and neurites might be remains unknown. Indeed, it is not clear whether it is actually the same protein that is accumulated at all three anatomical sites, or different site specific proteins. Certainly, the protein does not appear to be progranulin, since antibodies to this do not immunolabel any of the ubiquitinated structures present in patients with either chromosome-17 linked FTLD with PGRN mutations [1], or in patients within the present series with type 1 histology and PGRN mutations (where known), or in others with types 2 and 3 histology (Mann, unpublished data).
In conclusion, in this present study, we have described three distinct histological profiles that encompass the pathological entity of FTLD-U. We have been able to show that the various clinical representations of FTLD are strongly associated with each of the different histological subtypes. Each of these subtypes may have separate underlying pathophysiological causes which might reflect (1) different ubiquitinated proteins accumulating, (2) different forms of the same protein, or (3) different anatomical distributions of an identical protein, within each histological subtype. By refining our histological criteria of FTLD-U it might be possible to better determine genetic or other factors which cause or increase the risk of developing the different clinical or pathological forms of FTLD.
References
Baker M, Mackenzie IRA, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M (2006) Mutations in Progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442:916–919
Bergmann M, Kuchelmeister K, Schmid KW, Kretzschmar HA, Schroder R (1996) Different variants of frontotemporal dementia: a neuropathological and immunohistochemical study. Acta Neuropathol 92:170–179
Bigio EH, Johnson NA, Rademaker AW, Fung BB, Mesulam M-M, Siddique N, Dellefave L, Caliendo J, Freeman S, Siddique T (2004) Neuronal ubiquitinated intranuclear inclusions in familial and non-familial frontotemporal dementia of the motor neurone disease type associated with amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 63: 810–811
Brun A, Englund E, Gustafson L, Passant U, Mann DMA, Neary D, Snowden JS (1994) Clinical, neuropsychological and neuropathological criteria for fronto-temporal dementia. J Neurol Neurosurg Psychiatry 57:416–418
Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin J-J, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cujit I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C (2006) Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442:920–924
Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, Chatterjee A, Hurtig HI, Karlawish JH, Rosen HJ, Van Deerlin V, Lee V M-Y, Miller BL, Trojanowski JQ, Grossman M (2006) Frontotemporal dementia: clinicopathological correlations. Ann Neurol 59:952–962
Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, Jhaveri BS, Karlawish JH, Pestronk A, Smith TW, Tu P-H, Watts GDJ, Markesbery WR, Smith CD, Kimonis VE (2006) Novel ubiquitin pathology in frontotemporal dementia with valosin-containig protein gene mutations. J Neuropathol Exp Neurol 65:571–581
Froelich Fabre S, Axelman P, Almkvist A, Basun H, Lannfelt L (2003) Extended investigation of tau and mutation screening of other candidate genes on chromosome 17q21 in a Swedish FTDP-17 family. Am J Med Genet Part B (Neuropsychiatric Genet) 121B:112–118
Hodges JR, Davies R, Xuereb JH, Krill J, Halliday G (2003) Survival in frontotemporal dementia. Neurology 61:349–354
Hodges JR, Davies RR, Xuereb JH, Casey B, Broe M, Bak TH, Kril JJ, Halliday GM (2004) Clinicopathological correlates in frontotemporal dementia. Ann Neurol 56:399–406
Josephs KA, Holton JL, Rossor MN, Godbolt AK, Osawa T, Strand K, Khan N, Al-Sarraj S, Revesz T (2004) Frontotemporal lobar degeneration and ubiquitin immunohistochemistry. Neuropathol Appl Neurobiol 30:369–373
Josephs KA, Petersen RC, Knopman DS, Boeve BF, Whitwell JL, Duffy JR, Parisi JE, Dickson DW (2006) Clinicopathologic analysis of frontotemporal and corticobasal degenerations and PSP. Neurology 66:41–48
Katsuse O, Dickson DW (2005) Ubiquitin immunohistochemistry of frontotemporal lobar degeneration differentiates cases with and without motor neurone disease. Alzheimer Dis Assoc Disord 19(suppl 1):S37–S43
Kovari E, Gold G, Giannakopoulos P, Bouras C (2004) Cortical ubiquitin-positive inclusions in frontotemporal dementia without motor neurone disease: a quantitative immunocytochemical study. Acta Neuropathol 108:207–212
Lipton AM, White CL III, Bigio EH (2004) Frontotemporal lobar degeneration with motor neuron disease-type inclusions predominates in 76 cases of frontotemporal degeneration. Acta Neuropathol 108: 379–385
Mackenzie IRA, Feldman H (2003) The relationship between extramotor ubiquitin-immunoreactive neuronal inclusions and dementia in motor neurone disease. Acta Neuropathol 105:98–102
Mackenzie IRA, Shi J, Shaw CL, Du Pleassis D, Neary D, Snowden D, Mann DMA (2006) dementia lacking distinctive histology (DLDH) revisited. Acta Neuropathol (in press)
Mackenzie IR, Baker M, West G, Woulfe J, Qadi N, Gass J, Cannon A, Adamson J, Feldman H, Lindholm C, Melquist S, Pettman R, Sadovnick AD, Dwosh E, Whiteheart SW, Huton M, Pickering-Brown SM (2006) A family with tau-negative dementia and neuronal intranuclear inclusions linked to chromosome 17. Brain 129:853–867
Mann DMA (2005) The genetics and molecular pathology of frontotemporal lobar degeneration. In: Burns A, O’Brien J, Ames D (eds) Dementia, 3rd edn. Hodder Arnold, London, pp 689–701
Mott RT, Dickson DW, Trojanowski JQ, Zhukareva V, Lee VM, Forman M, Van Deerlin V, Ervin JF, Wang DS, Schmechel DE, Hulette CM (2005) Neuropathologic, biochemical, and molecular characterization of the frontotemporal dementias. J Neuropathol Exp Neurol 64:420–428
Neary D, Snowden JS, Mann DMA (2005) Frontotemporal dementia. Lancet Neurology 4:771–779
Neary D, Snowden JS, Mann DMA, Northen B, Goulding PJ, MacDermott N (1990) Frontal lobe dementia and motor neurone disease. J Neurol Neurosurg Psychiatr 53:23–32
Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF (1998) Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology 51:1546–1554
Rademakers R, Cruts M, Dermaut B, Sleegers K, Rosso SM, Van Den Broeck M, Backhovens H, van Swieten JC, van Duijn CM, van Broeckhoven C (2002) Tau-negative frontal lobe dementia at 17q21: significant finemapping of the candidate region to a 4.8-cm interval. Mol Psychiatry 7:1064–1074
Rosso SM, Kamphorst W, de Graaf B, Willemsen R, Ravid R, Niermeijer MF, Spillantini MG, Heutink P, van Swieten JC (2001) Familial frontotemporal dementia with ubiquitin positive inclusions is linked to chromosome 17q21–22. Brain 124:1948–1957
Rossor MN, Revesz T, Lantos PL, Warrington EK (2000) Semantic dementia with ubiquitin-positive tau-negative inclusion bodies. Brain 123: 267–276
Schroder R, Watts GDJ, Mehta SG, Evert BO, Broich P, Fliessbach K, Pauls K, Hans VH, Kimonis V, Thal DR (2005) Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol 57:457–461
Shi J, Shaw CL, Richardson AMT, Bailey K, Tian J, Varma AR, Neary D, Snowden JS, Mann DMA (2005) Histopathological changes underlying frontotemporal lobar degeneration with clinicopathological correlation. Acta Neuropathol 110:501–512
Snowden JS, Neary D, Mann DMA (1996) Fronto-temporal lobar degeneration: fronto-temporal dementia, progressive aphasia, semantic dementia. Churchill, Livingstone, pp 1–227
Taniguchi S, McDonagh AM, Pickering-Brown SM, Umeda Y, Iwatsubo T, Hasegawa M, Mann DMA (2004) The neuropathology of frontotemporal lobar degeneration with respect to the cytological and biochemical characteristics of tau protein. Neuropathol Appl Neurobiol 30:1–18
Wightman G, Anderson VER, Martin J, Swash M, Anderton BH, Neary D, Mann DMA, Luthert P, Leigh PN (1992) Hippocampal and neocortical ubiquitin-immunoreactive inclusions in amyotrophic lateral sclerosis with dementia. Neurosci Lett 139: 269–274
Woulfe J, Kertesz A, Munoz D (2001) Frontotemporal dementia with ubiquitinated cytoplasmic and intranuclear inclusions. Acta Neuropathol 102:94–102
Acknowledgment
We wish to acknowledge the technical assistance and expertise of Bev Dupuis, Lisa Laurence, Margaret Luk and Carmen Michelsen, the immunohistochemistry laboratory technologists at Vancouver General Hospital, and that of Richard Matthias and Roslyn Hall, the Neuropathology laboratory technicians at Hope Hospital and Newcastle General Hospital, respectively. Funding support for Dr. Mackenzie was provided by the Canadian Institutes of Health Research (CIHR) grant no. 74580. Dr Pickering-Brown is the recipient of a Medical Research Council (MRC) New Investigator Award, and receives other funding from MRC and the Motor Neurone Disease Association. The work of the Manchester Brain Bank is supported in part by The Alzheimers Research Trust, who also assisted this collaborative work between Manchester and Newcastle Alzheimers Research Trust Network Centres. The work of the Newcastle Brain Tissue Resource is supported by the Medical Research Council, Newcastle University and Newcastle upon Tyne Hospitals NHS Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mackenzie, I.R.A., Baborie, A., Pickering-Brown, S. et al. Heterogeneity of ubiquitin pathology in frontotemporal lobar degeneration: classification and relation to clinical phenotype. Acta Neuropathol 112, 539–549 (2006). https://doi.org/10.1007/s00401-006-0138-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0138-9