
Abstract
Introduction
Enlarged parietal foramina are variable ossification defects in the parietal bones that present as symmetric radiolucencies on skull radiographs. In contrast to the normal small parietal foramina, enlarged parietal foramina are a hereditary condition and genes associated with it have been identified.
Methods
A literature review was performed to discuss the many known findings related to enlarged parietal foramina.
Conclusions
Even though they remain asymptomatic in the majority of cases, they may be associated with other pathologies and occasionally become symptomatic. This article provides a comprehensive review of the current knowledge of enlarged parietal foramina.
Similar content being viewed by others
Introduction
Enlarged parietal foramina (EPF) are variable intramembranous ossification defects of the parietal bones and were first described in 1707, and received little attention until the 1940s [7, 14] (Figs. 1–3, 4, and 5). Alternate terms in the literature referring to this condition are fenestrae parietales symmetricae, foramina parietalia permagna, and giant parietal foramina [21]. Since its first description in the scientific literature the understanding of this condition has evolved substantially. In normal fetuses, the frontal, parietal, and squamous parts of the temporal bones undergo intramembranous ossification, which is a direct ossification of the vascularized membrane. These parts are usually ossified in the fifth month of gestation. When there is insufficient ossification around the parietal notch, they end up as large permanent foramina [25]. EPF are foramina located in the upper posterior angle of the parietal bone close to the intersection of the sagittal and lamboid sutures and present as symmetric, paired radiolucencies on skull radiographs [25]. The diameter of EPF can range in size from a few millimeters to several centimeters. A prevalence of 1:15,000 to 1:50,000 has been reported [14, 21, 25]. The condition is distinguished from small parietal foramina, which are approximately 1–2 mm in diameter, occur uni- or bilateral, and are variable in number. These are considered normal common variants of the parietal bones, allow for passage of emissary veins, and occur in about 60–70 % of the population [17, 21] and are believed to have no correlation with instances of small parietal foramina, and in one study there has been evidence of individuals with both small parietal foramina and enlarged at the same time [4, 23]. The hereditary nature of EPF and associated genes have been characterized [7]. In young children, this condition may present as a persistently enlarged posterior fontanelle caused by a single large central parietal bone defect, termed cranium bifidum [25]. During the first few years of life as calvarial growth continues cranium bifidum tends to resolve into two distinct, large parietal foramina through an ossification of a midline bridge [13, 18]. Cranium bifidum is also referred to as the Catlin mark [17]. Most EPF are asymptomatic. However, craniofacial anomalies including cleft palate, myelomeningocele, and encephaloceles are rarely associated [17, 21]. Meningeal, cortical, and vascular malformations of the posterior fossa have also been reported to accompany the ossification defects and may predispose to epilepsy [25]. Individuals with EPF have experienced symptoms of violent headaches, vomiting, and intense pain on application of mild pressure to the unprotected cerebral cortex [14, 25]. Most people with EPF have a positive family history as the condition is inherited in an autosomal dominant fashion with high, but incomplete penetrance [25]. Mutations of either MSX2 or ALX4 genes are associated with enlarged parietal foramina and molecular genetic testing is clinically available [16, 25].

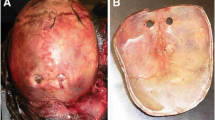
Adult calvaria with enlarged parietal foramina. Note that this is a misnomer as two of these specimens have normal parietal foramina

Coronal head CT noting the bilateral enlarged parietal foramina in a child with headache

Sagittal image of the patient in Fig. 4 noting the calvarial defect
Diagnosis
EPF and cranium bifidum may be diagnosed early in life during gestation or later incidentally as an adult [15, 25]. The defects are often palpable on exam as a flattened region or enlarged fontanelle posterior to the cranial apex. Radiographically a plain posterior–anterior skull film in an adult with EPF may coincidently demonstrate oval or round openings in the calvaria that resemble a “pair or spectacles”. On lateral view skull films they are more difficult to visualize since the lucencies are superimposed with normal bone. Fetal ultrasound can also detect EPF in a fetus at risk [25]. CT imaging with 3D reconstructions can delineate the osseous defect and MR imaging of the brain will demonstrate associated intracranial changes [25]. If EPF is associated with anomalies of the cerebral vasculature, additional vascular imaging like CT, MR, or digital subtraction angiography might be warranted [21]. In young children, EPF may present as a single aperture of the parietal bone, referred to as cranium bifidum as mentioned above [7, 25]. EPF is caused by a heterozygous mutation in the homeobox genes ALX4 and MSX2 located at 5q34–35 and 11p11, respectively [6]. EPF associated with either ALX4 or MSX2 mutations have a similar prevalence and are usually clinically indistinguishable [16]. Homeobox ALX4 and MSX2 genes encode a homeodomain, a protein structural domain that binds DNA or RNA, in transcription factors involved in skeletal development. A possible third locus on 4q21–q23 has also been reported in Chinese families [3]. Standard karyotyping and array comparative genomic hybridization are utilized to detect gross structural changes of MSX2 on 5q35.2 [1] or large deletions of ALX4 on 11p11.2 [26]. Molecular genetic testing is performed using sequence analysis and deletion/duplication analysis looking for sequence variants or whole gene/exon deletions, respectively [25]. On sequence analysis 6 ALX4 and 6 MSX2 mutations are identified in about 85 % of familial, but only 16 % of sporadic cases [16]. Except for multiple exostoses the frequency of mutation detection is generally lower if additional clinical abnormalities are present [25]. Prenatal diagnosis and preimplantation genetic testing during pregnancy is recommended for families with a known genetic defect associated with EPF at 18 to 20 weeks gestation [25].
Differential diagnosis
The differential diagnosis of EPF and cranium bifidum includes related disorders such as proximal 11 p deletion syndrome aka Potocki–Shaffer syndrome, ALX4-related frontonasal dysplasia, MSX2-related craniosynotosis, and distal 5q deletions [1, 25]. Potocki–Shaffer Syndrome, a contiguous gene deletion syndrome, is associated with loss of genetic material from chromosomal region 11p11.2p12. This syndrome’s abnormalities include intellectual disability, developmental delay, central nervous system abnormalities such as sensorineural hearing loss and visual problems, autistic behaviors, skeletal and craniofacial abnormalities, and abnormalities of the genitorurinary tract. The skeletal and craniofacial abnormalities include multiple exostoses and biparietal foramina [8, 12, 22]. The exostoses are associated with deletion of EXT2 and the biparietal foraminal are associated with ALX4 gene located proximal to EXT2. The genes related to the craniofacial abnormalities and mental retardation have not been identified. In addition, one familial case of Potocki–Shaffer Syndrome was unaffected by mental retardation [8]. ALX4-related frontonasal dysplasia is subtype of frontonasal dysplasia (FND), or median facial cleft and represents a failure to form the facial prominences around the primitive mouth. The genetic aspects of FND are not well defined. Aristaless-related homeobox genes like ALX1, ALX3, and ALX4 are thought to play a role in the development of structures derived from craniofacial mesenchyme, first branchial arch, and the limb bud [24, 26]. Two Turkish families, with histories of consanguinity, with a autosomal recessive inherited, homozygous nonsense mutation of ALX4 were described to have total alopecia, a large skull defect, coronal craniosynostosis, hypertelorism, agenesis of the corpus callosum, and mental retardation [10]. ALX4-related FND has also been associated with genital abnormalities [24]. Heterogeneous mutations in ALX4 were shown to cause cranium bifidum, but associated with a less severe phenotype compared to the autosomal recessive inheritance [26]. MSX2-related craniosynostosis is rare and has been found in a family with a single point mutation [5]. The same condition has also been associated with MSX2 duplications. Hypertelorism, growth and mental retardation, microcephaly, dysmorphic facial features such as downslanting palpebral fissures, prominent nasal bridge, strabismus, low set dysplastic ears, clinodactyly, brachydactyly, and heart defects are common features of this disorder [9]. Children with deletions of distal 5q can have defects of various systems from developmental delay to septal defects in the heart to cryptorchidism as well as a parietal foramina [1]. There have also been reports of scalp defects associated in one family with both enlarged parietal foramina and aplasia cutis congenita [20]. Parietal foramina can also appear as a feature of fetal methotrexate/aminopterin syndrome. Pregnant women that were previously exposed to folic acid antagonists in the first trimester as an attempt at medical abortion are at risk. In such cases there are wide spread deformities including syndactyly, hypodontia, cleft palate, and growth retardation and EPF [1, 2].
Treatment
Treatment for enlarged parietal foramina is generally conservative, but persistent cranium bifidum may warrant operative closure [25]. Associated seizures and headaches may be treated medically. Education of teachers and parents is advised to allay anxiety related to the disease and to eliminate anxiety about risk of penetrating injury to unprotected cerebrum [25]. Cranioplasty with autologous calvarial bone grafts or mesh plating systems with hydroxyapatite or methylmethacrylate is recommended for those at risk for injury, such as active young children and those with seizure disorders [11, 19]. Imaging to assess for accompanying venous anomalies is imperative prior to any surgical intervention [5].
Prognosis
Most EPF are asymptomatic and benign, but suspected associated abnormalities need to be considered [15]. Abnormal venous development including persistent falcine sinus with associated malformations of the straight sinus has been reported [5] (Figs. 6 and 7). These venous abnormalities are also encountered in atretic parietal encephaloceles raising the possibility of a common developmental origin [5]. Abnormalities of brain gyri have also been described including asymptomatic mesial occipital polymicrogyria [5, 21]. There is no described correlation between the size of the defect and the likelihood of having an associated brain abnormality [5]. A spontaneous decrease of the defect with growth of the infant has been observed, but closure is frequently incomplete [11].

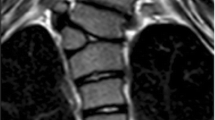
MRI of a 16-year-old female patient presenting with seizures. Note the vertically oriented persistent fetal vein

Axial MRI of the brain of the patient seen in Fig. 6. Note the persistent vein at the arrow tip
Conclusions
The so-called enlarged parietal foramina is a misnomer as many of these patients will also have normally sized parietal foramina. Some patients may present with symptoms and corrective surgery can be used. A strong genetic predisposition exists. These patients may have underlying venous abnormalities.
References
Aftimos S, Asquith P, Ashton F, Vasilevski O, Love DR (2010) Distal 5q deletion with associated parietal foramina. Clin Dysmorphol 19:43–47
Bawle EV, Conard JV, Weiss L (1998) Adult and two children with fetal methotrexate syndrome. Teratology 57:51–55
Chen G, Zhang D, Feng G, Liu W, He L (2003) A novel locus for parietal foramina maps to chromosome 4q21–q23. J Hum Genet 48:420–424
Currarino G (1976) Normal variants and congenital anomalies in the region of the obelion. AJR Am J Roentgenol 127:487–494
Fink AM, Maixner W (2006) Enlarged parietal foramina: MR imaging features in the fetus and neonate. AJNR Am J Neuroradiol 27:1379–1381
Ghassibe M, Bernier V, Boon LM, Vikkula M (2006) A novel mutation in the MSX2 homeobox gene of a family with foramina parietalia permagna, headache and vascular anomaly. Eur J Pediatr 165:734–735
Halbertsma T (1940) Fenestrae Parietales Symmetricae. Arch Dis Child 15:115–120
Hall CR, Wu Y, Shaffer LG, Hecht JT (2001) Familial case of Potocki-Shaffer syndrome associated with microdeletion of EXT2 and ALX4. Clin Genet 60:356–359
Kariminejad A, Kariminejad R, Tzschach A, Ullmann R, Ahmed A, Asghari-Roodsari A, Salehpour S, Afroozan F, Ropers HH, Kariminejad MH (2009) Craniosynostosis in a patient with 2q37.3 deletion 5q34 duplication: association of extra copy of MSX2 with craniosynostosis. Am J Med Genet A 149A:1544–1549
Kayserili H, Uz E, Niessen C, Vargel I, Alanay Y, Tuncbilek G, Yigit G, Uyguner O, Candan S, Okur H, Kaygin S, Balci S, Mavili E, Alikasifoglu M, Haase I, Wollnik B, Akarsu NA (2009) ALX4 dysfunction disrupts craniofacial and epidermal development. Hum Mol Genet 18:4357–4366
Kortesis B, Richards T, David L, Glazier S, Argenta L (2003) Surgical management of foramina parietalia permagna. J Craniofac Surg 14:538–544
Levenson D (2010) New information, recommendations for Potocki-Shaffer syndrome. Am J Med Genet A 152A: fmx
Little BB, Knoll KA, Klein VR, Heller KB (1990) Hereditary cranium bifidum and symmetric parietal foramina are the same entity. Am J Med Genet 35:453–458
Lodge T (1975) Developmental defects in the cranial vault. Br J Radiol 48:421–434
Luker J, McNamara T, Sandy J (1998) Holes in the head: parietal foramina, a developmental anomaly seen during a routine orthodontic assessment. Int J Paediatr Dent 8:209–211
Mavrogiannis LA, Taylor IB, Davies SJ, Ramos FJ, Olivares JL, Wilkie AO (2006) Enlarged parietal foramina caused by mutations in the homeobox genes ALX4 and MSX2: from genotype to phenotype. Eur J Hum Genet 14:151–158
Mupparapu M, Binder RE, Duarte F (2006) Hereditary cranium bifidum persisting as enlarged parietal foramina (Catlin marks) on cephalometric radiographs. Am J Orthod Dentofacial Orthop 129:825–828
Pang D, Lin A (1982) Symptomatic large parietal foramina. Neurosurgery 11:33–37
Perlyn CA, Schmelzer R, Govier D, Marsh JL (2005) Congenital scalp and calvarial deficiencies: principles for classification and surgical management. Plast Reconstr Surg 115:1129–1141
Preis S, Engelbrecht V, Lenard HG (1995) Aplasia cutis congenita and enlarged parietal foramina (Catlin marks) in a family. Acta Paediatr 84:701–702
Reddy AT, Hedlund GL, Percy AK (2000) Enlarged parietal foramina: association with cerebral venous and cortical anomalies. Neurology 54:1175–1178
Swarr DT, Bloom D, Lewis RA, Elenberg E, Friedman EM, Glotzbach C, Wissman SD, Shaffer LG, Potocki L (2010) Potocki-Shaffer syndrome: comprehensive clinical assessment, review of the literature, and proposals for medical management. Am J Med Genet A 152A:565–572
Tubbs RS, Smyth MD, Oakes WJ (2003) Parietal foramina are not synonymous with giant parietal foramina. Pediatr Neurosurg 39:216–217
Uz E, Alanay Y, Aktas D, Vargel I, Gucer S, Tuncbilek G, von Eggeling F, Yilmaz E, Deren O, Posorski N, Ozdag H, Liehr T, Balci S, Alikasifoglu M, Wollnik B, Akarsu NA (2010) Disruption of ALX1 causes extreme microphthalmia and severe facial clefting: expanding the spectrum of autosomal-recessive ALX-related frontonasal dysplasia. Am J Hum Genet 86:789–796
Wilkie AOM, Mavrogiannis LA (1993) Enlarged parietal foramina/cranium bifidum. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) GeneReviews™ [Internet]. Seattle (WA), University of Washington
Wuyts W, Cleiren E, Homfray T, Rasore-Quartino A, Vanhoenacker F, Van Hul W (2000) The ALX4 homeobox gene is mutated in patients with ossification defects of the skull (foramina parietalia permagna, OMIM 168500). J Med Genet 37:916–920
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Griessenauer, C.J., Veith, P., Mortazavi, M.M. et al. Enlarged parietal foramina: a review of genetics, prognosis, radiology, and treatment. Childs Nerv Syst 29, 543–547 (2013). https://doi.org/10.1007/s00381-012-1982-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-012-1982-7