
Abstract
In studying the regulation of GSH11, the structural gene of the high-affinity glutathione transporter (GSH-P1) in Saccharomyces cerevisiae, a cis-acting cysteine responsive element, CCGCCACAC (CCG motif), was detected. Like GSH-P1, the cystathionine γ-lyase encoded by CYS3 is induced by sulfur starvation and repressed by addition of cysteine to the growth medium. We detected a CCG motif (−311 to −303) and a CGC motif (CGCCACAC; −193 to −186), which is one base shorter than the CCG motif, in the 5′-upstream region of CYS3. One copy of the centromere determining element 1, CDE1 (TCACGTGA; −217 to −210), being responsible for regulation of the sulfate assimilation pathway genes, was also detected. We tested the roles of these three elements in the regulation of CYS3. Using a lacZ-reporter assay system, we found that the CCG/CGC motif is required for activation of CYS3, as well as for its repression by cysteine. In contrast, the CDE1 motif was responsible for only activation of CYS3. We also found that two transcription factors, Met4 and VDE, are responsible for activation of CYS3 through the CCG/CGC and CDE1 motifs. These observations suggest a dual regulation of CYS3 by factors that interact with the CDE1 motif and the CCG/CGC motifs.
Similar content being viewed by others

Introduction
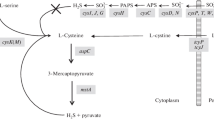
Genetic control of methionine biosynthesis has long been of interest (Thomas and Surdin-Kerjan 1997). Researchers in the de Robichon-Szulmajster Laboratory conducted an intensive study in Saccharomyces cerevisiae of methionine biosynthesis (Masselot and de Robichon-Szulmajster 1975), but its relationship to cysteine biosynthesis has remained obscure. S. cerevisiae can grow using methionine, homocysteine, cysteine, or glutathione as the sole sulfur source (Ono et al. 1984, 1988; Cherest and Surdin-Kerjan 1992). Subsequent work has determined that S. cerevisiae can reduce sulfate to form sulfide which is utilized to synthesize homocysteine (the sulfate assimilation pathway), and homocysteine-S is then converted to cysteine-S through cystathionine-S (the reverse trans-sulfuration pathway) (Ono et al. 1999). The reverse trans-sulfuration pathway consists of two enzymes, cystathionine β-synthase (βCTSase; EC 4.2.1.21) encoded by CYS4 and cystathionine γ-lyase (γCTLase; EC 4.4.1.1) encoded by CYS3.
In addition, we identified a high-affinity glutathionine transport system (GSH-P1) encoded by GSH11/HGT1/OPT1 in S. cerevisiae (Bourbouloux et al. 2000; Hauser et al. 2000; Miyake et al. 1998), and demonstrated that GSH11 is induced by sulfur starvation and repressed by addition of cysteine to the culture medium through a 5′-upstream sequence (CCGCCACAC), hereafter referred as the CCG motif (Miyake et al. 2002). In the course of that study, we identified VDE and seven additional proteins that bind to the CCG motif. The homing endonuclease VDE, which is produced by processing of the TFP1/VMA1 gene product (Hirata et al. 1990; Kane et al. 1990), is thought to function as a transcriptional activator of GSH11 because GSH11 is not expressed in a VDE-deleted strain, and the observed inability to express GSH11 is overcome by the introduction of the coding region of VDE or the entire VMA1 gene (Miyake et al. 2003). Given the observation that the activity of γCTLase increases markedly under sulfur starvation and decreases markedly when cells are grown in the presence of organic sulfur compounds such as glutathione, methionine, and cysteine (Ono et al. 1991), we decided to investigate the regulation of CYS3.
In examining the 5′-upstream region of CYS3, we detected a CCG motif (CCGCCACAC) together with a CGCCACAC sequence, which is one nucleotide shorter than the CCG motif, and hereafter referred to as the CGC motif. In addition, a CDE1 motif (TCACGTGA) which is found in centromeres and in the 5′-upstream regions of genes encoding enzymes of the sulfate assimilation pathway (Barton et al. 1993; Kuras and Thomas 1995) was detected. A helix–loop–helix protein encoded by CBF1 binds to the CDE1 motif and forms a heteromeric complex with Met4 and Met28 at the MET16 promoter (Kuras and Thomas 1996). In addition, a cbf1 null mutant displays methionine auxotrophy (Baker and Masison 1990). Genes in the sulfate assimilation pathway are regulated coordinately and MET4 is indispensable for their expression (Thomas and Surdin-Kerjan 1997). Given the presence of these three putative regulatory elements, we hypothesized that CYS3 would exhibit regulatory features similar to the sulfate assimilation pathway genes on the one hand and to GSH11/HGT1/OPT1 on the other. In this study, we focused on the following three putative regulatory motifs in CYS3: CCG (CCGCCACAC, −311 to −303), CDE1 (TCACGTGA, −217 to −210) and CGC (CGCCACAC, −193 to −186) (Fig. 1). Our results reveal the complex regulatory control of S. cerevisiae CYS3, and we discuss our findings in relation to S. cerevisiae sulfur metabolism.

Comparison of the 5′-upstream regions of CYS3, GSH11, and MET17. The cysteine-responsive element (−364 to −356; CCG) of GSH11, and the two regulatory centromere determining elements (−326 to −319, −301 to −294; CDE1) of MET17. The three putative regulatory motifs of CYS3 are shown as CCG (−311 to −303), centromere determining element (−217 to −210; CDE1), and CGC (−193 to −186), respectively
Materials and methods
Strains and plasmids
The S. cerevisiae strains used in this study were SF1-1C (MATa leu2 trp1 met17) and YPH500 (MATα ade2 lys2 his3 leu2 trp1 ura3), which are wild-type with respect to γCTLase activity and regulation. In the YOC2176 [MATα ade2 lys2 his3 leu2 trp1 ura3 VMA1-101 (vde-delta)] strain, the VDE region has been disrupted (Miyake et al. 2003) and we constructed the YPH500m4 (MATα ade2 lys2 his3 leu2 trp1 ura3 met4::CgHIS3) strain, by disrupting MET4 in the YPH500 strain. The Escherichia coli strain DH10B was used for amplification of plasmids. We used p69-2-1 (Ono et al. 1992) as a template for PCR amplification of CYS3, pBluescriptII SK+ (TOYOBO, Osaka, Japan) for sequencing, pMC1587 (containing the lacZ coding region (Casadaban et al. 1983)) for analysis of the CYS3 promoter region and pCgHIS3 (supplied by Dr. Harashima) for disruption of the MET4 gene.
Growth media and growth conditions
We used standard yeast growth media (Sherman et al. 1986) such as YPD (1% yeast extract, 2% peptone, and 2% glucose) and synthetic minimal (SD) medium. Sulfur-free (SF) medium was prepared by replacing the sulfate salts in the SD medium (Wickerham 1956) with the corresponding chloride salts (Ono et al. 1991). Where appropriate, supplements were added to the SD and SF media. For example, MET and CYS media were prepared by the addition of 100 μM methionine and 100 μM (derepression) or 10 mM (repression) cysteine. Escherichia coli was cultivated in LB broth (1% tryptone, 0.5% yeast extract, and 0.5% NaCl, pH 7.2) (Sambrook et al. 1989), with the addition of 50 μg/ml ampicillin when required. Solid media contained 2% agar. Saccharomyces cerevisiae and E. coli were grown at 30 and 37°C, respectively, and liquid cultures were incubated on a reciprocal shaker at 120 rpm.
DNA manipulations
DNA manipulation (Sambrook et al. 1989) and transformation of S. cerevisiae (Ito et al. 1983) were performed as described previously.
Disruption of MET4
A DNA fragment containing the Candida glabrata HIS3 gene was amplified by PCR from pCgHIS3 using the primers: 5′-AGT CCC ACG AAG GCG ACT CAT ACA GCA CGG AAT TCA TAA AGT TGT AAA ACG ACG GCC AGT-3′ and 5′-ATG GAG CTT AGA AAA GAA GCC TCT GCT ACT ACA CCG TGC TCA CAG GAA ACA GCT ATG ACC-3′. The italicized sequences correspond to 10 bp downstream from the MET4 ATG initiation codon and 951 bp upstream from the TAG termination codon, respectively. PCR-amplification was performed for one cycle at 94°C for 1 min, 56°C for 30 s, and 1 min at 72°C, 30 cycles at 94°C for 30 s, 60°C for 30 s, and 1 min at 72°C, with a final extension step at 72°C for 7 min. The YPH500 strain was transformed with the 1.78 kb PCR product and His+ Met− transformants were obtained. We confirmed that the transformant YPH500m4 was met4 − by complementation with the MET4-bearing plasmid pYCM4 (provided by Dr. Omura, Institute for Liquor Products, Suntory Ltd.).
Electrophoretic mobility shift assays
Saccharomyces cerevisiae SF1-1C was grown overnight in YPD and then transferred to either SF or fresh YPD media. After a 17 h incubation, the cells were harvested by centrifugation, washed once with homogenization buffer (20 mM Tris–HCl, pH 8.0, 1 mM EDTA, 1 mM dithiothreitol (DTT), 10% glycerol, 1 mM phenylmethylsulphonyl fluoride, 1 mM benzamidine–HCl) and resuspended in 0.3 ml of the same buffer with 0.4 ml glass beads. The cell suspension was subjected to ten cycles of shaking for 30 s and cooling in an ice bath for 60 s; cell debris was then removed by centrifugation. The supernatant was used as the cell extract.
The mobility shift assays were performed as described previously (Miyake et al. 2002). In brief, DNA–protein binding reactions used cell extract (20 μg of protein) and digoxigenin-11-ddUTP (DIG)-labeled DNA probes (100 fmol) in a total volume of 10 μl containing 10 mM HEPES (pH 7.6), 1 mM EDTA, 10 mM (NH4)2SO4, 1 mM DTT, 1% (w/v) Tween 20, 1 μg poly(dI–dC) and 30 mM KCl. Reactions were performed at 25°C for 30 min and terminated by adding 2 μl of loading buffer containing 0.2× TBE buffer and 40% glycerol. Fragments-1, -2, -3, -6, -7, and -8 were produced by PCR using the primers listed in Table 1 and end-labeled with DIG using terminal transferase, according to the manufacturer’s instructions (Roche Diagnostics GmbH, Mannheim, Germany). Two DIG-labeled fragments (F-4 and F-5) were custom-synthesized (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Using 2% agarose gels that had been pre-run (2 h) in 0.5× TBE buffer, terminated reaction mixtures were subjected to electrophoresis for 2 h at 4°C, and then blotted onto nylon membranes (Amersham Pharmacia Biotech) by capillary transfer. DIG-labeled DNA was detected using an immunoassay with NBT/BCIP as the color-developing substrate, according to the manufacturer’s protocol (Roche Diagnostics).
Competition assays
Competition experiments were performed as described above, with the addition of 100 pmol unlabeled oligonucleotides as the competitor. Four unlabeled double stranded fragments (URM-1, URM-2, URM-3, and URM-4) were custom-synthesized (Amersham Pharmacia Biotech).
Construction of plasmids for the lacZ reporter assay
Using p69-2-1 and the primers listed in Table 1, we PCR-amplified various fragments containing different segments of the 5′-upstream region of CYS3. The nucleotide sequence of the reverse primer (USC3-rev) encoded 12 amino acids from the N terminus of γCTLase and a BamHI site was designed near the 5′ end. This reverse primer was used in conjunction with forward primers to various segments of the 5′-upstream region of CYS3. Forward primers contained a SmaI site at the 5′ end. Although most PCR fragments were digested with SmaI and BamHI, and then ligated into the SmaI/BamHI site of pMC1587, fragments F20, F22 and F23 were digested with SmaI and ligated into the SmaI site in the F19 plasmid (Fig. 5). All gene constructs encoded proteins in which the first eight amino acids of β-galactosidase were replaced by the first 12 amino acids of γCTLase.
Assay for β-galactosidase activity
Cells were incubated in CYS (SF medium supplemented with 10 mM cysteine) or SF media for 17 h, then harvested and resuspended in 1 ml of Z buffer (Sherman et al. 1986), followed by treatment with 0.05 ml chloroform and 0.05 ml 0.1% SDS. β-galactosidase activity was measured at 30°C as the mean rate of hydrolysis of o-nitrophenyl-β-d-galactopyranoside (ONPG) to o-nitrophenol (ONP) and galactose. β-galactosidase activity was calculated using the formula A420 × 1,000 min−1 ml−1 OD −1600 .
Results
Electrophoretic mobility shift assays using segments from the 5′-upstream region of CYS3
To determine whether the transcriptional activators VDE and Met4 bind to the 5′-upstream region of CYS3, a 368-bp DNA fragment containing the three putative binding motifs CCG, CDE1, and CGC (Fig. 2a) was used as a probe (fragment F-1) in electrophoretic mobility shift assays. The probe DNA was mixed with cell extracts prepared from a wild-type strain (YPH500), a met4 mutant (met4::CgHIS3), or a vde-delta strain. In order to examine the effects of sulfur starvation, the strains were grown in the presence (YPD and SD) or absence (SF) of sulfate. As shown in Fig. 2b, regardless of the presence or absence of sulfate in the growth medium, all strains showed a slow-migrating band, indicating that all strains constitutively produced protein(s) that bound to the probe. However, the electrophoretic mobility of the band differed markedly between SD medium and SF medium for the wild-type and Δmet4 strains, whereas such differences were not observed for the Δvde strain. This result is consistent with our previous observation that γCTLase activity increases in cells grown in SF medium but not YPD or SD medium (Ono et al. 1991).

Electrophoretic mobility shift assay using cell extracts from cells grown under various conditions. a The fragment indicated (F-1) was amplified using the primers listed in Table 1 (USC3-1 and USC3-rev). PCR product was end-labeled with DIG using terminal transferase. b The DIG end-labeled DNA fragment containing all three putative motifs (CCG, CDE1, and CGC) was added to the reaction mixtures in the presence of cell extracts from three kinds of strains [YPH500 (wild-type), YPH500m4 (met4::CgHIS3), and YOC2176 (vde-delta)] grown in the presence (YPD and SD) or absence of sulfur (SFree)
Next, various segments of the 5′-upstream region of CYS3 were PCR-amplified (Fig. 3a) and subjected to electrophoretic mobility shift assays in the presence of extracts prepared from wild-type cells grown in SF medium (Fig. 3b). The fragments F-2 (−73 to +36) and F-3 (−185 to +36) containing one and two TATA boxes, respectively, exhibited substantial band shifts in the presence of the cell extract, consistent with previous observations that S. cerevisiae possesses protein(s) that binds to the TATA box including the TATA-binding protein (TBP) (Buratowski et al. 1988). The fragment F-4 (−203 to −164), which contained a CGC motif, exhibited a marked band shift. The fragment F-5 (−233 to −194), which contained a CDE1 motif, revealed more than one shifted band, indicating a complex interaction of this fragment with different proteins. The fragments F-6 (−302 to −223) and F-7 (−302 to −241), which correspond to intervening segments between the CCG and CDE1 motifs, showed substantial band shifts, indicating the presence of protein-binding site(s) in this region. The fragment F-8 (−332 to −241) corresponding to F-7 plus the CCG motif exhibited a weaker band shift than the F-7 fragment. This result indicates that the binding of protein(s) to the CCG motif is to some extent inhibited by the presence of the neighboring nucleotide sequence. Taken together, these results show that S. cerevisiae grown under sulfur starvation conditions expresses a number of proteins that bind to the three motifs of interest.

Electrophoretic mobility shift assay. a The five fragments indicated (F-2, -3, -6, -7, -8) were amplified using the primers listed in Table 1 (F-2: USC3-6 and USC3-rev; F-3: USC3-8 and USC3-rev; F-6: USC3-3 and USC3-12; F-7: USC3-3 and USC3-11; and F-8: USC3-1 and USC3-11). PCR products were end-labeled with DIG using terminal transferase. The two DIG end-labeled DNA fragments indicated (F-4 and F-5) were custom-synthesized. b The DIG end-labeled DNA fragments containing consensus sequences from the CYS3 promoter region were added to the reaction mixtures in the presence (+) or absence (−) of SFree-grown cell extract. The location of the putative regulatory motifs are as shown in Fig. 1
In order to investigate the protein-binding sites in the F-4 (CGC motif) and F-5 (CDE1 motif) fragments, we performed competition assays using different sequences from the 5′ upstream region of CYS3 (Fig. 4a). URM-1 (−219 to −170; lane 3) and URM-3 (−193 to −174; lane 5) caused marked suppression of the band-shift of F-4, whereas URM-2 (−203 to −184; lane4) and URM-4 (−185 to −170; lane 6) had no effect (Fig. 4b). We therefore concluded that protein(s) bind not to the CGC motif alone but to the CGC motif plus its 3′ downstream sequence. In contrast, URM-1 and URM-2 were highly and only slightly suppressive, respectively, to proteins binding the F-5 fragment, suggesting that protein(s) bind to the CDE1 motif alone.

Competition assay. a The six custom-synthesized fragments indicated [URM1–URM4, DIG end-labeled F-4 (CGC), and DIG end-labeled F-5 (CDE1)] correspond to different segments of the 5′-upstream region of CYS3. The fragments corresponding to −203 to −164 (CGC) and −233 to −194 (CDE1) were used as the target DNA and the other fragments were used as competitors. The locations of the putative regulatory motifs are as shown in Fig. 1. b In the left panel, the target CGC fragment was incubated alone or with cell extract (lanes 1 or 2) in the absence of competitor, or in the presence of a 1,000-fold excess of unlabeled double stranded URM1 (lane 3), URM2 (lane 4), URM3 (lane 5), or URM4 (lane 6). In the right panel, the target CDE1 fragment was incubated alone or with cell extract (lanes 7 or 8) in the absence of competitor, or in the presence (lanes 9 and 10) of a 1,000-fold excess of the unlabeled competitor as indicated
lacZ reporter assays
γCTLase activity increases when cells are grown in SF medium (Ono et al. 1991). We therefore decided to determine the regulatory motifs involved in transcriptional activation of CYS3, under sulfur starvation conditions, with a lacZ reporter assay using 5′-upstream segments of CYS3. We constructed plasmids in which various 5′-upstream segments of CYS3 were inserted in front of lacZ (Fig. 5). The constructs were then introduced into the YPH500 strain (wild-type), and β-galactosidase activity was measured (Table 2, 1st column). Although 100 μM cysteine in the growth medium caused a decrease in γCTLase activity in our previous study (Ono et al. 1991), 100 μM or even 1 mM cysteine, did not cause a decrease in β-galactosidase activity. The F07 construct exhibited β-galactosidase activity of 611.4 U min−1 OD −1600 in the presence of 1 mM cysteine. However, 10 mM cysteine caused an obvious decrease in β-galactosidase activity. We attribute the observed difference in the effective cysteine concentration to the inhibitory effect of cysteine on γCTLase but not on β-galactosidase. Therefore, we used 10 mM cysteine for the subsequent studies. In the absence of cysteine, the F07 construct (containing all three motifs of interest) exhibited β-galactosidase activity of 617.4 U min−1 OD −1600 , whereas in the presence of 10 mM cysteine, the activity level was 309.1 U min−1 OD −1600 , indicating that cysteine caused a 50% decrease in gene expression relative to the derepressed level. The F17 construct (containing one TATA box) exhibited a β-galactosidase activity of 2.9 U min−1 OD −1600 regardless of the presence or absence of cysteine. The F19 (containing two TATA boxes) and F22 (containing the CDE1 motif in addition to two TATA boxes) constructs exhibited approximately ninefold and 81-fold the activity, respectively, of the F17 construct regardless of the presence or absence of cysteine. However, the fold induction by F22 was approximately the same as by F19 and F17. Therefore, we concluded that the CDE1 element is involved in constitutive expression of the target gene but not in sulfur starvation-dependent expression.

Plasmids used for the lacZ reporter assay. In order to construct plasmids containing sequential deletions, we PCR-amplified various DNA fragments from p69-2-1 using the primers indicated. These fragments contained different combinations of the CCG, CDE1, and CGC motifs. The PCR products were digested with SmaI and BamHI, then ligated into the SmaI/BamHI site of pMC1587. The internal deletion plasmids containing CCG and/or CDE1 were generated by PCR amplification using Pyrobest DNA polymerase (Takara, Shiga, Japan). The fragments F20 (CCG motif), F22 (CDE1 motif) and F23 (CCG and CDE1 motifs) were digested with SmaI, and ligated into the SmaI site of F19
In contrast, the F18 construct (containing the CGC motif in addition to the two TATA boxes) exhibited 80% the activity of the F07 construct in the absence of cysteine, and 30% the activity of F07 in the presence of 10 mM cysteine, indicating that the CGC motif is responsible for strong activation in the absence of cysteine, and strong repression in its presence (5-fold induction). Since the F21 construct (containing the CCG motif located in the position of the CGC motif) behaved similarly to the F18 construct, it is possible that, in closer proximity to the coding region, the CCG motif can function in a similar manner to the CGC motif (2.9-fold induction). The F20 construct (containing the CCG motif and its adjacent downstream regions) exhibited approximately half the activity of the F21 construct in the absence of cysteine. Notably, the F20 construct contains not only the CCG motif but also the adjacent regions. Therefore, we concluded that either the sequence(s) adjacent to the CCG motif are inhibitory or the distance between the motif and its target gene is critical for the activator function of the CCG motif.
As observed in comparison of the F10, F08, and F23 constructs, the presence of the CDE1 motif plus either the CGC motif or the CCG motif caused a high level of constitutive expression of the target gene. In contrast, the CCG and CGC motifs alone are responsible for a high level of inducible expression.
Involvement of the homing nuclease VDE in the regulation of CYS3
To ascertain the involvement of VDE in regulation of CYS3, lacZ reporter constructs were introduced into the YOC2176 strain (a vde-delta derivative of YPH500; Table 2, 2nd column). In the absence of cysteine, F07 (containing all three motifs of interest) exhibited somewhat lower activity in the Δvde mutant (YOC2176) than in the wild-type strain (YPH500); that is, the fold induction by F07 was 2× for the wild-type strain and 1.5× for the Δvde mutant. In the presence of cysteine, there was no detectable difference between the two strains. A more significant difference was observed for the F18 (CGC motif) and F21 (CCG motif) constructs. In the absence of cysteine, both constructs exhibited approximately one-third the activity in the Δvde mutant as in the wild-type strain. In contrast, there was no detectable difference between the strains in the presence of cysteine. F22 (CDE1 motif) caused slightly lower gene expression in the Δvde mutant, relative to the wild-type strain, in the absence of cysteine. F10 showed approximately two-thirds the level of gene expression in the Δvde mutant relative to the wild-type strain in the absence of cysteine, while both strains exhibited similar levels of gene expression in the presence of 10 mM cysteine. Therefore, VDE may play a role in activation of CYS3. In the absence of cysteine, similar β-galactosidase activities were observed with the F07, F10, F08, and F23 constructs. The latter three constructs contain the CDE1 motif plus either the CGC (F10 and F08) or CCG motif (F23); the level of gene expression due to these constructs was reduced by the addition of cysteine. In contrast, the F18 (CGC motif alone), F20 and F21 (CCG motif alone), and F22 (CDE1 motif alone) constructs induced intermediate levels of gene expression, and the expression level was not affected by the addition of cysteine. Hence, the expression of CYS3 via the CGC motif or the CCG motif is weaker in the Δvde mutant than in the wild-type strain.
These results suggest the following conclusions: (1) the CGC and CCG motifs are equally effective for CYS3 activation, and this activation is not subjected to repression by cysteine; (2) the CDE1 motif alone exhibits a level of activation similar to the CGC and CCG motifs; and (3) the CGC and CCG motifs increase activation in the presence of the CDE1 motif, and this activation is subject to repression by cysteine. The wild-type strain (YPH500) and the vde-delta strain (YOC2176) exhibited a marked difference in activation mediated by the CGC (F18) or CCG (F21) motif and by combining the CDE1 motif with either the CGC (F10 and F08) or CCG (F23) motif. Based on the present observations, together with our previous finding that VDE binds the CCG motif (Miyake et al. 2003), we contend that VDE plays a role in activation of CYS3 via the CGC and CCG motifs.
Role of MET4 in the regulation of CYS3
In order to investigate the role of MET4 in regulation of CYS3, we constructed the met4 mutant YPH500m4 and performed lacZ reporter assays using this strain (Table 2, the 3rd column). In all assays, the level of β-galactosidase activity was low, suggesting that MET4 is required for regulation mediated by each of the three motifs. However, the F22 construct (CDE1 motif alone) exhibited a higher level of induction in the met4 mutant (YPH500m4) than in the wild-type strain (YPH500); that is, the fold induction for construct F22 was 0.9× for the wild-type strain and 1.9× for the met4 mutant. This result suggests that an auxiliary transcription factor other than Met4 is involved in expression of CYS3 under sulfur starvation conditions.
Discussion
In this study, we examined the roles of three putative regulatory motifs present in the 5′-upstream region of the S. cerevisiae CYS3 gene, CGC (−193 to −186), CDE1 (−217 to −210), and CCG (−311 to −303). We found that the CCG and CGC motifs play roles in expression of the target gene and that differences observed between the motifs are largely attributable to their proximity to the target gene. As the CGC motif (CGCCACAC) is only one nucleotide shorter than the CCG motif (CCGCCACAC), we suggest that the functional body of the motifs is identical and should be referred to as the CCG/CGC motif. Activation by the CCG/CGC motif is repressed by 10 mM cysteine, and this response is lowered substantially in the absence of VDE, suggesting that VDE binds directly or indirectly to the CCG/CGC motif to activate the target gene. We have also found that a high concentration of cysteine (10 mM) is necessary for repression of the CYS3 gene. Our result is consistent with the results reported by Hansen and Johannesen (2000), in that cysteine repression of the sulfate assimilation pathway genes MET14 and MET25 occurred at a concentration of 10 mM cysteine. Thus, quite high levels of cysteine appear to be needed for repression of the CYS3, MET14, and MET25 genes.
The CDE1 motif is responsible for intermediate activation of the target gene, but not for repression by cysteine. Activation via the CDE1 motif and via the CCG/CGC motif was additive, indicating that distinct activation complexes are interacting with these two kinds of motif. Activation due to the CDE1 motif appears to be dependent upon VDE, though to a much lesser extent than the VDE dependence of the CCG/CGC motif. Furthermore, it is clear that MET4 is required for activation via both the CDE1 motif and the CCG/CGC motif. In view of these and earlier findings, we propose a model whereby CYS3 is under dual regulation by the CDE1 motif and the CCG/CGC motif (Fig. 6). We suggest that both the Cbf1–Met4 and VDE–Met4 complexes bind to the CDE1 motif and the CCG/CGC motif, and that these two regulatory systems can operate independently as well as cooperatively. As binding of TBP to the TATA box serves as an anchor for RNA polymerase to bind DNA for the initiation of RNA synthesis (Buratowski et al. 1988), we hypothesize that TBP binds directly or indirectly to the Cbf1–Met4 and VDE–Met4 complexes.

A proposed model for regulation of CYS3. In the absence of cysteine, the transcription factor complexes Met4–Cbf1 and/or Met4–VDE bind the CDE1 or CCG/CGC motifs, respectively, then these complexes interact with basal transcription factors such as TBP, resulting in the activation of CYS3 transcription. In the presence of cysteine, CYS3 expression is repressed by the abundant cysteine molecules interacting directly or indirectly with VDE
In this investigation, we focused on the CCG/CGC and CDE1 regulatory motifs. However, CYS3 also possesses a Met31/Met32 binding site (MBS motif; −198 to −193) (Blaiseau et al. 1997; Chiang et al. 2006). Our data (Table 2) indicate that the CCG/CGC motif regulates expression of CYS3, but the MBS motif does not. In the 5′-upstream region of CYS3, there are also regulatory motifs that appear to be involved in the stress response, such as the yAP-1 response element (YRE motif; −235 to −229) (Lee et al. 1999) and the Xbp1 binding site (XBS motif; −166 to −161) (Mai and Breeden 2000). Since glutathione plays a central role in the cellular response to oxidative stress and is synthesized using cysteine as a substrate, there appears to be a link between expression of CYS3 and the roles of these stress response motifs. This line of investigation remains for future studies.
It is now clear that the CCG/CGC motif is responsible for regulation of both GSH11 and CYS3. By searching the S. cerevisiae genome (http://www.yeastgenome.org/), we found that several ORFs have motifs identical or similar to the CCG/CGC motif in their 5′-upstream regions. While some are involved in sulfur metabolism, such as SPE2 (encoding SAM decarboxylase, EC 4.1.1.50), MET13 (encoding a putative 5,10-methylenetetrahydrofolate reductase, EC 1.5.1.20), and MET2 (encoding homoserine O-acetyltransferase, EC 2.3.1.31) (Fig. 7), the functions of the others are not known. It will be of interest in future studies to determine whether or not these genes and ORFs are regulated via Met4 and VDE.

Regulation of S. cerevisiae sulfur metabolism by cysteine. The boxed sulfur metabolism genes contain one or more CCG/CGC motif(s) in their 5′-upstream regions. CYS3 and GSH11 are regulated by the amount of available cysteine in the cell
References
Baker RE, Masison DC (1990) Isolation of the gene encoding the Saccharomyces cerevisiae centromere-binding protein CP1. Mol Cell Biol 10:2458–2467
Barton AB, Kaback DB, Clark MW, Keng T, Oouellette BF, Storms RK, Zeng B, Zhong W, Fortin N, Delaney S, Bussey H (1993) Physical localization of yeast CYS3, a gene whose product resembles the rat γ-cystathionase and Escherichia coli cystathionine γ-synthase enzymes. Yeast 9:363–369
Blaiseau P-L, Isnard A-D, Surdin-Kerjan Y, Thomas D (1997) Met31p and Met32p, two related zinc finger proteins, are involved in transcriptional regulation of yeast sulfur amino acid metabolism. Mol Cell Biol 17:3640–3648
Bourbouloux A, Shahi P, Chakladar A, Delrot S, Bachhawat AK (2000) Hgt1p, a high affinity glutathione transporter from the yeast Saccharomyces cerevisiae. J Biol Chem 275:13259–13265
Buratowski S, Hahn S, Sharp PA, Guarente L (1988) Function of a yeast TATA element-binding protein in a mammalian transcription system. Nature 334:37–42
Casadaban MJ, Martinez-Arias A, Shapira SK, Chou J (1983) Beta-galactosidase gene fusions for analyzing gene expression in Escherichia coli and yeast. Methods Enzymol 100:293–308
Cherest H, Surdin-Kerjan Y (1992) Genetic analysis of a new mutation conferring cysteine auxotrophy in Saccharomyces cerevisiae: updating of the sulfur metabolism pathway. Genetics 130:51–58
Chiang DY, Nix DA, Shultzaberger RK, Gasch AP, Eisen MB (2006) Flexible promoter architecture requirements for coactivator recruitment. BMC Mol Biol 7:16
Hansen J, Johannesen PF (2000) Cysteine is essential for transcriptional regulation of the sulfur assimilation genes in Saccharomyces cerevisiae. Mol Gen Genet 263:535–542
Hauser M, Donhardt AM, Barnes D, Naider F, Becker JM (2000) Enkephalins are transported by a novel eukaryotic peptide uptake system. J Biol Chem 275:3037–3041
Hirata R, Ohsumk Y, Nakano A, Kawasaki H, Suzuki K, Anraku Y (1990) Molecular structure of a gene, VMA1, encoding the catalytic subunit of H+-translocating adenosine triphosphatase from vacuolar membranes of Saccharomyces cerevisiae. J Biol Chem 265:6726–6733
Ito H, Fukuda Y, Murata K, Kimura A (1983) Transformation of intact yeast cells treated with alkali cations. J Bacteriol 153:163–168
Kane PM, Yamashiro CT, Wolczyk DF, Neff N, Goebl M, Stevens TH (1990) Protein splicing converts the yeast TFP1 gene product to the 69-kD subunit of the vacuolar H+-adenosine triphosphatase. Science 250:651–657
Kuras L, Thomas D (1995) Identification of the yeast methionine biosynthetic genes that require the centromere binding factor 1 for their transcriptional activation. FEBS Lett 367:15–18
Kuras L, Cherest H, Surdin-Kerjan Y, Thomas D (1996) A heteromeric complex containing the centromere binding factor 1 and two basic leucine zipper factors, Met4 and Met28, mediates the transcription activation of yeast sulfur metabolism. EMBO J 15:2519–2529
Lee J, Godon C, Lagniel G, Spector D, Garin J, Labarre J, Toledano MB (1999) Yap1 and Skn7 control two specialized oxidative stress response regulons in yeast. J Biol Chem 274:16040–16046
Mai B, Breeden L (2000) CLN1 and its repression by Xbp1 are important for efficient sporulation in budding yeast. Mol Cell Biol 20:478–487
Masselot M, de Robichon-Szulmajster H (1975) Methionine biosynthesis in Saccharomyces cerevisiae. I. Genetical analysis of auxotrophic mutants. Mol Gen Genet 139:121–132
Miyake T, Hazu T, Yoshida S, Kanayama M, Tomochika K, Shinoda S, Ono B (1998) Glutathione transport systems of the budding yeast Saccharomyces cerevisiae. Biosci Biotechnol Biochem 62:1858–1864
Miyake T, Kanayama M, Sammoto H, Ono B (2002) A novel cis-acting cysteine-responsive regulatory element of the gene for the high-affinity glutathione transporter of Saccharomyces cerevisiae. Mol Genet Genomics 266:1004–1011
Miyake T, Hiraishi H, Sammoto H, Ono B-I (2003) Involvement of the VDE homing endonuclease and rapamycin in regulation of the Saccharomyces cerevisiae GSH11 gene encoding the high-affinity glutathione transporter. J Biol Chem 278:39632–39636
Ono B, Suruga T, Yamamoto M, Yamamoto S, Murata K, Kimura A, Shinoda S, Ohmori S (1984) Cystathionine accumulation in Saccharomyces cerevisiae. J Bacteriol 158:860–865
Ono B, Shirahige Y, Nanjoh A, Andou N, Ohue H, Ishino-Arao Y (1988) Cysteine biosynthesis in Saccharomyces cerevisiae; mutation that confers cystathionine β-synthase deficiency. J Bacteriol 170:5883–5889
Ono B, Naito K, Shirahige Y, Yamamoto M (1991) Regulation of cystathionine γ-lyase in Saccharomyces cerevisiae. Yeast 7:843–848
Ono B, Tanaka K, Naito K, Heike C, Shinoda S, Yamamoto S, Ohmori S, Oshima T, Toh-e A (1992) Cloning and characterization of the CYS3 (CYI1) gene of Saccharomyces cerevisiae. J Bacteriol 174:3339–3347
Ono B, Hazu T, Yoshida S, Kawato T, Shinoda S, Brzvwczy J, Paszewski A (1999) Cysteine biosynthesis in Saccharomyces cerevisiae: a new outlook on pathway and regulation. Yeast 15:1365–1375
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sherman F, Fink GR, Hicks JB (1986) Laboratory course manual for methods in yeast genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor
Thomas D, Surdin-Kerjan Y (1997) Metabolism of sulfur amino acids in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 61:503–532
Wickerham LJ (1956) A critical evaluation of the nitrogen assimilation tests commonly used in the classification of yeast. J Bacteriol 52:293–301
Acknowledgments
This work was supported by funds from the Bio-Venture project and the 21st century COE program of Ritsumeikan University. We are grateful to S. Harashima (Osaka University) and F. Omura (Institute for Liquor Products, Suntory Ltd) for their kind gifts of plasmids containing Candida glabrata HIS3 and MET4, respectively.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by K. Kuchler.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hiraishi, H., Miyake, T. & Ono, Bi. Transcriptional regulation of Saccharomyces cerevisiae CYS3 encoding cystathionine γ-lyase. Curr Genet 53, 225–234 (2008). https://doi.org/10.1007/s00294-008-0181-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-008-0181-2