
Abstract
Autophagy is a ubiquitous catabolic process that ensures organism’s well-being by sequestering a wide array of undesired intracellular constituents into double-membrane vesicles termed autophagosomes for lysosomal degradation. Interest in autophagy research has recently gained momentum as it is increasingly being recognized to play fundamental roles in diverse aspects of human pathophysiology including virus infection and its subsequent complications. This review discusses recent advances in autophagy studies with respect to virus infection and pathogenesis. A growing body of evidence suggests that the autophagy pathway and/or autophagy genes play pleiotropic functions in the host’s intrinsic, innate, and adaptive immune response against viruses. However, some viruses have evolved to encode virulence factors that evade or counteract the execution of autophagy. Furthermore, certain viruses are equipped to enhance autophagy or exploit the autophagy machinery for their replication and pathogenesis. A comprehensive understanding of the roles of autophagy pathway and autophagy genes during viral infection may enable the discovery of novel antiviral drug targets.
Similar content being viewed by others

Introduction
For humans, one’s “well-being” is all about maintaining a balance in every aspect of life. The same principle applies to virtually all eukaryotic organisms’ well-being, and one of the most appropriate representations of this in scientific terms would be “autophagy,” meaning self-eating in Greek. Eukaryotic cells have evolutionarily acquired this self-cannibalization process to ensure a balance between biosynthesis and biodegradation such that cellular homeostasis is sustained during unfavorable conditions like nutrient deprivation. With the ability to adapt to a variety of metabolic stresses, the autophagic machinery serves as a cellular scavenger of undesired intracellular constituents, directing its cargo to the lysosomal compartment for degradation and ultimately, recycling. There are three types of autophagy, defined by their physiological functions and the mode of cargo delivery to the lysosome: macroautophagy, microautophagy, and chaperone-mediated autophagy, with macroautophagy (hereafter referred to as autophagy) being the most well-characterized type in eukaryotic cells. This catabolic process via autophagy is executed through a unique membrane trafficking event that generates double-membrane vesicles, termed autophagosomes. Autophagosomes in mammalian cells were first observed through electron microscopy in the 1950s [1], but autophagy was then merely considered to be a cellular garbage disposal system. However, the past decade has been an era of enormous research on autophagy, disclosing the integral roles of autophagy in human physiology such as adaptation to neonatal starvation during development, structural remodeling during cell differentiation, and life span extension during ageing. Moreover, an increasing body of research is recently revealing the protective roles of autophagy against pathogenesis of human diseases, including cancer, neurodegeneration, cardiovascular and liver disease, myopathies, and infectious disease [2]. This review primarily focuses on the role of autophagy pathway and/or autophagy genes as a host defense mechanism during intrinsic, innate, and adaptive immune responses to microbial infections, especially viruses. Furthermore, this review also highlights how viruses have developed strategies that subvert and/or hijack the host autophagic machinery to benefit their propagation.
Molecular mechanisms of autophagy
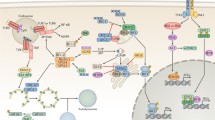
Autophagy is a bona fide cytoprotective sensor that removes any unwelcomed intracellular content including reactive oxygen species (ROS), long-lived or aggregated proteins/lipids, damaged organelles, and invading microbes. A low level of basal autophagy occurs constitutively to maintain cellular homeostasis by controlling the quality and routine turnover of intracellular entities. Upon sensing stimuli, however, it is rapidly up-regulated as cytoplasmic components are enwrapped by a de novo formed double-membrane crescent called the phagophore. The two extremities of the elongating phagophore, also termed as isolation membrane, are sealed to form a closed double-membrane vacuole, the autophagosome. The autophagosome subsequently matures by fusing with the lysosome to form a single membrane vesicle, named the autolysosome, where the encapsulated contents together with the inner membrane of the autophagosome undergo breakdown by lysosomal hydrolases. Eventually, the resulting macromolecular building blocks—amino acids, fatty acids, and nucleotides—are exported to the cytoplasm for recycling. Although the molecular regulation of the autophagy pathway is presently the subject of vigorous investigation by a number of laboratories, it is yet far from complete. There are currently over 30 autophagy genes (ATG) identified in yeast, and the corresponding human ATG homologs have been characterized in higher eukaryotes as well. The autophagy gene products form specific complexes in four distinct stages of autophagosome biogenesis—induction, vesicle nucleation, vesicle elongation, and vesicle fusion (Fig. 1).

The molecular regulation of the autophagy pathway. Over 30 autophagy gene products form distinct complexes during four sequential stages of autophagy: induction, nucleation of the phagophore, elongation and completion of the autophagosome membrane, and fusion of the autophagosome with lysosome for degradation of the sequestered cargoes. a mTOR plays a central role in constitutively suppressing induction of autophagy by binding and phosphorylating the ULK1/2-Atg13-FIP200 complex. Upon autophagy stimulation, including nutrient deprivation or rapamycin treatment, mTOR is repressed and thus causes hypophosphorylation of the ULK1/2-Atg13-FIP200 complex, leading to the induction of autophagy. b Formation of the phagophore is mediated by the class III PI3K complex, which consists of three major components, including Vps34, Vps15, and Beclin1 (yellow). The activity of this lipid kinase complex depends on various positive (green) and negative (red) regulators that associate with Beclin1. c Growth and closure of the autophagosome membrane involves two ubiquitin-like conjugation systems, Atg12 and LC3, which associate with the phagophore and promote its curvature and expansion. d The completed autophagosome fuses with lysosome and matures into autolysosome by the action of various positive (green) and negative (red) regulators. Consequently, the inner membrane and the luminal constituents of the autolysosome are degraded by lysosomal acid hydrolases and exported to the cytoplasm for reuse
Autophagy induction
In order for a basal level of autophagy to persist at low levels when nutrients are ample, the induction of autophagy is tightly suppressed by a number of regulators. A central inhibitor of autophagy induction is the mammalian target of rapamycin (mTOR), a serine/threonine protein kinase which functions as an ATP and amino acid sensor to balance nutrient availability and cell growth through the regulation of cap-dependent translation. Under nutrient-replete conditions, mTOR is in its active conformation and represses autophagy by phosphorylating Unc-51-like kinase 1 and 2 (ULK1/2) (mammalian homologue of yeast Atg1) and Atg13, which triggers the disassembly of the ULK1/2-Atg13-FIP200 complex (Atg1-Atg13-Atg17 complex in yeast). However, upon nutrient depletion, class I PI3K/Akt signaling becomes attenuated, relieving the repressive effect of mTOR on autophagy. Consequently, hypo-phosphorylated Atg13 associates with ULK1/2 and up-regulates ULK kinase activity in the ULK1/2-Atg13-FIP200 complex [3], leading to autophagy induction by facilitating anterograde transport of downstream molecules to phagophores. However, not all autophagy signals are transduced through the class I PI3K/Akt/mTOR pathway upon nutrient depravation. In response to a wide variety of stimuli, the initiation of the autophagic cascade can also be regulated by other molecules such as 5′-AMP-activated protein kinase (AMPK), phosphorylated eukaryotic initiation factor 2α (eIF2α), p53, c-jun-N-terminal kinase 1 (JNK1), inositol-requiring enzyme-1 (IRE-1), inositol-triphosphate receptor (IP3R), and intracellular calcium [2].
Vesicle nucleation
Vesicle nucleation refers to the assembly of phagophores, and the major complex responsible for this step is the lipid class III phosphatidylinositol 3-kinase (C3-PI3K) complex consisting of three core components: Vps34, Vps15 (formerly called p150), and Beclin1 (Atg6 in yeast). Vps34 is the enzymatic subunit that phosphorylates phosphatidylinositol (PtdIns) to generate phosphatidylinositol 3-phosphate (PI3P), and its specific activity significantly depends on a regulatory subunit Vps15, a myristoylated serine/threonine kinase [4]. The consequence of PI3P localization at phagophores in mammalian cells needs to be further elucidated, but it has been reported that the localized production of PI3P by Vps34 enables proteins containing FYVE and PX domains to be recruited to specific membrane compartments [5]. As such, the yeast C3-PI3K complex is critical for recruiting autophagy proteins to the preautophagosomal structure (PAS), a site of autophagosome formation in yeast [6]. Beclin1, which is known to be a potent haploid-insufficient tumor suppressor, plays a fundamental role in integrating multiple signals that positively or negatively regulate autophagy. Although it does not exhibit any enzymatic activity alone, Beclin1 serves as a platform for the recruitment of a variety of regulators of the C3-PI3K complex. Beclin1-binding proteins that activate the C3-PI3K complex include Atg14-like protein [Atg14L, also called Beclin1-associated autophagy-related key regulator (Barkor)], UV irradiation resistance-associated gene (UVRAG), Bax-interacting factor-1 (Bif-1) or Endophillin B1, and activating molecule in Beclin-1-regulated autophagy protein-1 (Ambra-1) [7–11]. Conversely, Bcl-2/BclxL and Run domain protein as Beclin1 interacting and cysteine-rich containing (Rubicon) associate with Beclin1 to repress vesicle nucleation and the autophagosomal maturation activity of C3-PI3K complex, respectively [7, 12].
Vesicle elongation
Two evolutionarily conserved ubiquitin-like conjugation systems, Atg12 and LC3/Atg8, are indispensible for the elongation of autophagic vesicles. The first system involves the covalent association of Atg12 to Atg5 by the sequential action of E1-like enzyme Atg7 and E2-like enzyme Atg10. Furthermore, Atg5 binds to a coiled-coil domain-containing protein, Atg16L1, to form a heterotrimeric complex. Although phagophore formation is initially independent of the presence of the Atg5–Atg12–Atg16L1 complex during its early stage, the complex associates with the expanding phagophore—mostly on the outer surface—and dissociates from the membrane upon completion of autophagosome formation. The second system entails the conjugation of microtubule-associated protein 1A/1B, light chain 3 (LC3) (mammalian homologue of yeast Atg8) to phosphatidylethanolamine (PE). After cleavage by the cysteine protease Atg4, soluble LC3 (LC3-I) is converted to a lipidated form (LC3-PE or LC3-II) as PE is added to the exposed glycine residue on the carboxyl terminus of the cleaved LC3. This conjugation is executed by E1-like enzyme Atg7, followed by E2-like enzyme Atg3, and the Atg5–Atg12–Atg16L1 complex provides an E3-like activity [13]. The resulting LC3-PE stably associates with both the outer and inner membranes of the expanding phagophore. Although the precise role of the LC3-PE conjugate remains obscure, it has been reported that Atg8 in Saccharomyces cerevisiae mediates the tethering and hemifusion of liposomes in response to lipidation in vitro, suggesting that Atg8 plays a vital role in the enlargement of the autophagosomal membrane [14]. In line with this, mammalian LC3 is essential for autophagosome formation, presumably by providing the driving force for the deformation or curvature of the expanding membrane. Unlike Atg5–Atg12 conjugation, LC3 lipidation is reversible: LC3-PE on the outer membrane of the completed autophagosome gets cleaved by Atg4 to release free LC3. However, LC3-PE on the inner membrane of autophagosome is retained, thus enabling the utilization of membrane-embedded LC3 as a marker for autophagosomes through immunoblotting or microscopic analyses. Additionally, it should be noted that an adaptor protein p62, also known as SQSTM1/sequestome 1, is widely used as a marker to monitor the extent of autophagic degradation, as the total cellular protein level of p62 is inversely correlated with autophagic flow. This is because p62 is involved in selective autophagy of mono- or poly-ubiquitinated protein aggregates by directing them to LC3-PE embedded in the surface of autophagosomes, wherein it, along with the recruited proteins, undergoes lysosomal degradation.
The components of the two ubiquitin-like conjugation systems have been considered to be essential for the formation of autophagosomes. However, surprisingly, an alternative Atg5/Atg7-independent macroautophagy pathway has been recently discovered in mammalian cells [15]. Mouse embryonic fibroblasts (MEFs) lacking Atg5 or Atg7 were still capable of forming autophagosomes and autolysosomes without the lipidation of LC3. Instead, they performed autophagy-mediated protein degradation in a Rab9-dependent manner. Moreover, unlike conventional Atg5/Atg7-dependent autophagy where autophagic membranes stem, at least in part, from thin type (6–7 nm) such as endoplasmic reticulum (ER), the alternative autophagy generated autophagosomes through the fusion of isolation membranes with vesicles derived from thick type (9–10 nm) including trans-Golgi or late endosomes. The Atg5/Atg7-independent autophagy was triggered under certain stress conditions in vitro and in the clearance of organelles during erythroid maturation in vivo, suggesting differential roles of the alternative autophagy pathway. Yet, more in-depth studies are required to elaborate mechanistic basis for this alternative autophagy pathway and its physiological relevance.
Vesicle fusion
Despite recent advances in the research on the autophagy pathway, the molecular basis underlying autophagosomal fusion to lysosomes has remained largely enigmatic. In yeast, the machinery is comprised of soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) proteins including Vam3, Vam7, Vti1, and Vkt6; members of class C vacuolar protein sorting (Vps) complex, especially homotypic vacuole fusion and protein sorting (HOPS) complex; Rab family GTPase Ypt7; and three other proteins such as Sec18, Ccz1, and Mon1 [16]. The molecular machinery involved in autophagosomal maturation in mammalian cells is even more poorly understood. The lysosomal membrane protein LAMP-2, the small GTPase Rab7 (mammalian homologue of yeast Ypt7), AAA ATPase SKD1, Hrs, SNARE protein Vti1, and the UVRAG-containing C3-PI3K complex in coordination with the HOPS complex have been characterized to mediate autophagosomal fusion to lysosomes [17–21]. These observations suggest that by sharing similar machinery, autophagic maturation may be interconnected with the endocytic trafficking pathway for concomitant lysosomal fusion. However, this represents only the tip of the iceberg, and more comprehensive studies on the molecular regulation of autophagosomal fusion to endosomes or lysosomes are necessary.
Autophagy as host defense machinery
Autophagy has been evolutionarily developed to be part of the host defense system as it contributes to the discarding of invading microorganisms by delivering them to the lysosomal compartment. Such degradation activity against any non-self, rather than self, entities via an autophagy-related process is coined as “xenophagy” [22]. The ability of xenophagy to allow an intrinsic elimination of intruding viruses, bacteria, fungi, or parasites makes it one of the most central immune players during a microbial infection. In addition, autophagy pathway and/or autophagy genes have been shown to be crucial for a successful innate and adaptive immune response upon a pathogen encounter. This encompasses signaling pathways involved with eIF2α, Toll-like receptors (TLRs), interferon-γ (IFN-γ), and p47 GTPase. Moreover, autophagy genes have been characterized to participate in the processing and presentation of antigen onto major histocompatibility (MHC) molecules, lymphoid development and survival, and thymic selection [23]. Although it is not yet clear whether the antimicrobial defense functions of autophagy genes are irrespective of the autophagy pathway itself, increasing number of autophagy proteins, including Beclin1, Atg5, Atg7, Atg8/LC3, and Atg16L1, have been reported to function in immune pathways. This idea may not be entirely unprecedented as previous studies have revealed independent and alternative roles of autophagy genes in other cellular processes such as apoptosis, membrane trafficking, and axonal elongation [24]. Recent studies provide a growing body of evidence regarding pleiotropic roles of autophagy in antiviral immunity (Fig. 2).

Crosstalk between host autophagy and viruses. a The autophagy pathway functions as an intrinsic antiviral defense mechanism by directly engulfing virions and/or viral components of HSV-1 or SIN for lysosomal degradation, an activity known as xenophagy. b Autophagy can participate in homeostatic regulation of innate immune signaling against VSV. The autophagic machinery transfers RNA replication intermediates of VSV from the cytosol to the endosomal compartment containing TLR7, promoting the production of type I IFNs in pDCs. However, the autophagy pathway and/or autophagy genes may suppress RLR signaling and the production of type I IFNs in MEFs and primary macrophages either by suppressing the activities of RIG-1 and IPS-1 in MEFs or by reducing the intracellular level of ROS associated with dysfunctional mitochondria in primary macrophages. c Autophagy also functions in antiviral adaptive immunity by facilitating the processing and presentation of viral antigens onto MHC molecules. Upon EBV or Influenza A virus infection, autophagosomes target antigens derived either from incoming virions or endogenously expressed viral proteins to MHC class II loading compartment (MIIC). Notably, nuclear membrane-derived, four-layered autophagosomes can target endogenous viral antigens for MHC class I presentation during HSV-1 infection. d On the flip side, some viruses subvert and, moreover, enhance the host autophagic machinery for replication and pathogenesis. Several herpesviruses have been shown to block induction, nucleation, or elongation of autophagic vesicles. Several RNA viruses such as poliovirus and CVB3, HIV-1, and HCV induce the formation of vesicles resembling autophagosomes to enhance viral replication or non-lytic egress. However, it is yet unclear whether these viruses lead to the accumulation of these autophagosome-like structures by blocking their fusion with lysosomes
Autophagy in intrinsic immunity (xenophagy)
Intrinsic immunity has been defined as “a collection of activities of constitutively expressed genes that fall outside the conventional definitions of innate and adaptive immune systems but nevertheless provide potent protection from viral infection” [25]. A number of studies has indicated that xenophagy is a novel type of intrinsic immunity as it instantly triggers the elimination of undesired foreign materials including viruses and their components. Xenophagy induced by viral infections can be perfectly exemplified by Beclin1, the first mammalian autophagy protein identified. Discovered as a Bcl-2 interaction protein from a yeast-two hybrid screening, Beclin1 was originally described in the context of the mouse central nervous system (CNS) infection with Sindbis virus (SIN), an enveloped positive-sense single-stranded RNA virus in the alphavirus genus of the Togaviridae family. Transmitted through mosquitoes, SIN causes only a mild fever and arthritis in humans, but it serves as a useful model for studying human alphavirus encephalomyelitis in mice. The brains of mice infected with a recombinant SIN chimera over-expressing full-length human Beclin1 had fewer SIN RNA-positive cells, fewer apoptotic cells, and lower viral titers than those of mice infected with SIN over-expressing human Beclin1 mutants lacking the Bcl-2-binding domain or containing a premature stop codon near the 5′ terminus, which eventually protected these mice against lethal encephalitis [26]. However, the molecular mechanism by which Beclin1 protects mice neurons from SIN infection has remained largely unanswered. It can be speculated that enforced expression of the Beclin1 may lead to the up-regulation of xenophagy of SIN. Alternatively, Beclin1 may reduce SIN-induced neuronal apoptosis through the up-regulation of xenophagy-dependent or xenophagy-independent mechanisms since committing suicide upon SIN infection may not be such a clever antiviral strategy for terminally differentiated neurons with limited mitotic capacity. Intriguingly, a recent study has revealed that the antiviral response induced by SIN infection is achieved by selective autophagy against viral structural proteins [27]. The authors demonstrate that the inactivation of Atg5 in SIN-infected mouse hippocampal neurons causes increased apoptosis and, astonishingly, co-accumulation of SIN capsid proteins and p62 without affecting the viral titer. Moreover, p62 was co-immunoprecipitated with the capsid protein and siRNA-mediated knockdown of p62 and an essential autophagic protein, Atg7, resulted in an augmentation of the steady-state level of the SIN capsid protein and virus-induced cell death. These observations indicate that neuronal protection against SIN is exerted through the clearance of viral capsid proteins via p62-mediated selective xenophagy, and that the failure to eliminate SIN capsid proteins induces neuronal apoptosis through uncharacterized mechanisms. Thus, it is tempting to speculate that the enforced expression of Beclin1 may contribute to neuronal protection against SIN by enhancing p62-mediated selective xenophagic activity, which dampens SIN replication in a non-self-destructive way. Moreover, a recent study illustrating a novel proapoptotic function of the Beclin1 carboxyl terminus after cleavage by caspase-8 [28] may allow us to exclude the possibility that Beclin1 possesses a specific function in reducing SIN-induced neuronal apoptosis in an autophagy/xenophagy-independent manner. However, further investigations are necessary to elaborate the mechanisms by which SIN capsid proteins induce cell death.
Xenophagy also contributes to the protection of mice and humans from fatal encephalitis caused by herpes simplex virus type 1 (HSV-1), a double-stranded DNA virus in the genus simplexvirus of the Alphaherpesviridae family. Upon reactivation from latency, HSV-1 causes a variety of clinical syndromes, ranging from mild mucocutaneous diseases such as cold sores to fatal encephalitis. Identification of the HSV-1 neurovirulence factor ICP34.5 was a major breakthrough in understanding fatal encephalitis in mice and humans. Studies in the past few years have found that ICP34.5 prevents protein kinase activated by double-stranded RNA (PKR)-mediated translational shutoff by recruiting protein phosphatase 1α (PP1α) to eIF2α for dephosphorylation, contributing to neurovirulence by antagonizing the antiviral function of PKR, which inhibits translational initiation by phosphorylating eIF2α [29, 30]. Since one of the consequences of PKR activation is the induction of autophagy, ICP34.5 thereby also hinders PKR/eIF2α-mediated autophagy activation. Accordingly, whereas induction of autophagy was suppressed in wild-type MEFs infected with wild-type HSV-1, autophagy activity was increased in wild-type MEFs infected with HSV-1 mutants lacking ICP34.5. This study further showed that this phenotype is abrogated in PKR-deficient MEFs, suggesting that PKR is essential for the induction of autophagy caused by infection with ICP34.5-deficient HSV-1. However, HSV-1 encoding ICP34.5 mutants that still retain their ability to release PKR-mediated translational shutoff were still avirulent in vivo [31, 32], indicating that ICP34.5 needs a second target to establish complete neurovirulence. In fact, it was later demonstrated that ICP34.5 targets Beclin1 through a direct interaction and that ICP34.5 mutant viruses deficient in Beclin1 binding caused increase in the number of autophagosomes in primary neurons [27]. These observations suggest that xenophagy degrades HSV-1 and that HSV-1 ICP34.5 subverts an early step of xenophagy, namely autophagosome formation, by targeting Beclin1 in addition to PKR. Moreover, ICP34.5 mutant virus that lost Beclin1 binding caused less mortality from HSV-1 encephalitis in wild-type mice, while restoring neurovirulence in PKR-deficient mice. These observations collectively suggest that xenophagy plays an essential role in protecting the mammalian central nervous system against HSV-1 pathogenesis and that PKR lies upstream of Beclin1 in the induction of autophagy pathway by HSV-1. Taken together, studies on SIN and HSV-1 acute pathogenesis imply that xenophagy is important for protection against CNS caused by both RNA and DNA neurotropic viruses.
Studies in plants have also demonstrated an essential role of autophagy-related genes in xenophagy-mediated viral clearance and homeostatic regulation of antiviral immune responses. Plants confer resistance when invading microbes trigger a type of program cell death (PCD) termed hypersensitive response (HR), which manifests as discrete necrotic lesions in otherwise phenotypically normal tissue [33]. Since they lack a phagocytosis-like system for cleaning up damaged or dead cells, plants have developed a novel mechanism that restricts HR PCD to occur exclusively at the site of infection to minimize unwanted cell death. It has been reported that autophagy-related genes in Arabidopsis function as negative regulators of HR PCD upon an encounter with tobacco mosaic virus (TMV), a positive-sense single-stranded RNA virus in the tobamovirus genus [34]. When BECLIN 1, PI3K/VPS34, ATG3, or ATG7 was silenced, TMV-induced HR PCD spread into adjacent healthy tissue as well as leaves distal to the infection site while initiation of HR PCD was intact. Moreover, GFP-LC3/Atg8 puncta and electron microscopic images of autophagic bodies demonstrated that autophagy is induced not only at the TMV infection site but also at uninfected adjacent tissue and distal uninfected leaves. These observations suggest that autophagy-related genes and autophagy pathway play a role in preventing pro-death signals from spreading to innocent bystander cells surrounding the HR PCD area. However, interestingly, autophagosomes were still readily detected 48 h after pathogen recognition, suggesting that the autophagic machinery may have additional roles in viral resistance. It is tempting to speculate that autophagy may participate in the eradication of HR PCD-derived cellular damage caused by nucleases, proteases, and hydrolases activated during antiviral defense response. Furthermore, xenophagic degradation of TMV may be another antiviral mechanism as knockdown of BECLIN1 also led to an increased accumulation of GFP-labeled TMV at the infection site without spreading outward. Further studies are necessary to elucidate the mechanisms by which autophagy prevents TMV replication and the propagation of pro-death signals during an antiviral response. Nontheless, these studies in the plant model organism provide further evidence that the antiviral function of autophagy is evolutionarily conserved.
Autophagy in innate immunity (TLR and RLR signaling)
Observations ranging from Drosophila to various types of mammalian cells have collectively suggested that autophagy and/or autophagy-related genes can also modulate multiple targets of innate immune responses to Vesicular stomatitis virus (VSV) infection in a cell type-specific fashion. VSV is a nonsegmented negative-sense single-stranded RNA virus in the vesiculovirus genus of the Rhabdoviridae family, and it is transmitted to arthropods and mammals. A recent study has demonstrated that autophagy plays a protective role VSV infection in Drosophila, in a manner analogous to TMV infection in plants with both organisms lacking adaptive immunity and relying only on their intrinsic and innate immune systems [35]. The study observed that double-stranded RNA (dsRNA)-mediated knockdown of numerous autophagy genes (Atg1, Atg2, Atg4, Atg5, Atg6, Atg7, Atg8b, Atg9, or Atg18) result in the elevation of VSV-infected cells in parallel with viral titer, implying that the antiviral autophagic machinery targets an early step, rather than a later step, of the VSV life cycle. Moreover, autophagic vacuoles were induced by UV-inactivated, replication-defective VSV to a similar extent as wild-type VSV, suggesting that a pre-existing molecule belonging to the incoming virus is sufficient to trigger the autophagic antiviral program. The study further claimed that, among the four components composing VSV virions (RNA genome, ribonucleoprotein complex, nucleocapsid, and glycoprotein), the surface glycoprotein of VSV (VSV-G) serves as a pathogen-associated molecular pattern for an as-of-yet undefined immune receptor at either the cell surface or endosomal compartment, which leads to the down-regulation of the autophagy suppressive insulin/PI3K/Akt/mTOR pathway. Yet, additional studies are required to elucidate the molecular basis of the recognition of VSV-G and subsequent induction of autophagy.
Several studies have indicated that the recognition of VSV-G by a pattern recognition receptor, such as TLRs, may trigger the xenophagic degradation of VSV. For example, VSV-G interacts with the TLR4 pathway in murine myeloid dendritic cells (mDCs) [36], TLR4 signaling can activate autophagy in human and murine macrophages [37], and the nine TLRs in Drosophila have been proposed as a pattern recognition receptor for the recognition of VSV-G [35]. On the other hand, autophagy mediates the activation of TLR signaling by delivering viral ligands to TLRs. For example, it has been reported that the autophagic machinery in plasmocytoid dendritic cells (pDCs) is necessary for delivering RNA replication intermediates of VSV and Sendai virus (SV) from the cytoplasm to the endosomal lumen where the single-stranded RNA (ssRNA) sensor, TLR7, resides [38]. Mature Atg5 −/− pDCs, which were obtained from irradiated wild-type mice reconstituted with liver and spleen cells from Atg5 −/− neonates, showed diminished secretion of interferon-α (IFN-α) and proinflammatory cytokine interleukin-12 (IL-12) upon VSV infection. Furthermore, VSV-infected bone marrow cells showed suppressed secretion in the presence of autophagy inhibitors such as wortmannin and 3-methyladenine (3-MA), which inhibit autophagosome formation without affecting endocytosis-mediated entry or the subsequent steps involved in VSV infection. It is possible that viral RNA is non-selectively captured by newly synthesized autophagosomes whose fusion with endosomes leads to the delivery of viral ligands to TLR7. Nevertheless, these observations definitively demonstrate the importance of autophagy in the innate recognition of viral pathogens by TLR7 and the subsequent production of IFN-α and IL-12. Collectively, a series of studies in recent years have revealed a cross-talk between the autophagic machinery and TLR signaling pathways as an antiviral innate immune response in that autophagy can exert a direct effect on the extermination of intracellular viruses following an activation of TLR signaling. Alternatively, autophagic trafficking can contribute to the activation of TLR signaling by transfering viral genetic materials to TLRs.
Somewhat paradoxically, several studies have demonstrated that autophagy-related genes in certain cell types can suppress the production of type I IFNs (IFN-α and IFN-β) and proinflammatory cytokines. Two distinct models have been proposed to explain the immunosuppressive role of autophagy and/or autophagy-related genes in innate immune responses against VSV. The first study showed that Atg5–Atg12 conjugates associate with two potent cytoplasmic dsRNA sensors of the RIG-I-like receptor (RLR) signaling, retinoic acid-inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5), and a mitochondrial adaptor protein named interferon-β promoter stimulator 1 (IPS-1) through their caspase recruitment domains (CARDs) to repress production of type I IFNs [39]. Atg5-deficient MEFs were resistant to VSV replication due to a robust production of type I IFNs in response to the unique dsRNA structure of ssRNA viruses (e.g., 5′-triphosphate of dsRNA in the case of RIG-I). Analogous phenotypes were observed in Atg7-deficient MEFs, in which the Atg5–Atg12 conjugation process is impaired. Notably, the interaction between Atg5 and IPS-1 was detected in the absence of VSV infection, as well, indicating that Atg5–Atg12 conjugate may suppress RLR signaling under normal physiological conditions to maintain cellular homeostasis. Despite how this first study clearly shows a non-canonical role of the Atg5–Atg12 conjugate in suppressing antiviral RLR signaling, an apparent conundrum lies in whether this observed phenotype results from a deficiency of the Atg5–Atg12 conjugate in the autophagy pathway or in an alternative, as-of-yet uncharacterized cellular mechanism that is independent of autophagy. Thus, it is important to examine whether the genetic ablation of additional autophagy genes that are indispensible for certain stages of autophagy pathway other than Atg5–Atg12 conjugation also permits the up-regulation of RLR signaling against VSV.
On the other hand, a second study observed that the accumulation of dysfunctional mitochondria in Atg5-deficient MEFs results in the hyperproduction of IFN-α and IL-6 and resistance to VSV infection through the up-regulation of RLR signaling in two ways [40]. Firstly, they showed that an elevated intracellular level of IPS-1 in accumulated dysfunctional mitochondria led to the amplification of RLR signaling. In addition, ROS, by-products of oxidative respiration in mitochondria, were largely responsible for the up-regulation of RLR signaling upon the accumulation of damaged mitochondria in Atg5-deficient MEFs because treatment with antioxidants—N-acetyl-l-cysteine (NAC) or propyl gallate (PG)—significantly dampened type I IFN and cytokine production in Atg5-deficient MEFs and primary macrophages. Moreover, chemically induced (Rotenone-treated) mitochondrial ROS was sufficient to potentiate RLR signaling in wild-type MEFs and macrophages, further corroborating the idea that ROS positively regulates RLR signaling. It can be concluded from these two completely different, but not necessarily mutually exclusive, models that autophagy-related genes and/or autophagy play an essential role in the homeostatic regulation of host innate antiviral defense by negatively regulating RLR signaling. However, further investigations are necessary to clarify whether the autophagic machinery indeed has a direct immunosuppressive function that augments susceptibility to viral infection or if it simply functions as an autoregulatory negative feedback loop to sophisticatedly coordinate inflammatory responses whose over-stimulation can otherwise lead to immunopathological consequences that are detrimental to the host.
Autophagy in adaptive immunity (antigen presentation)
The importance of autophagy’s role in adaptive immunity is highlighted by a recent study demonstrating that, in murine macrophages infected with HSV-1, autophagy facilitates viral glycoprotein B (gB) processing for antigen presentation on MHC class I, which leads to CD8+ T cell activation [41]. In the early phase of HSV-1 infection when ICP34.5 blocks the assembly of autophagosomes by targeting PKR and Beclin1, HSV-1 gB was processed through the classical pathway of MHC class I presentation by way of proteasomal degradation, translocation of the peptide into the ER lumen, and transport of the loaded MHC class I molecule to the cell surface. During the late phase of infection, however, a novel type of autophagosome characterized by four-layered, LC3-positive vesicles originating from both the inner and outer nuclear membranes, was formed. Interestingly, macrophages infected with an ICP34.5-depleted mutant virus rapidly generated classical double-layered autophagosomes and allowed vacuolar processing of gB antigens, beginning from the early stage of infection and persisting throughout the entire course of infection. These observations indicate that nuclear membrane-derived autophagy is favored when the classical autophagy pathway is inhibited, and that host cells respond to virus-induced suppression of autophagy by activating this newly discovered type of autophagy to facilitate MHC class I presentation for CD8+ T cell activation. Astonishingly, endogenous viral protein gB trapped by this unique autophagosome were subsequently hydrolytically degraded in the lumen of autolysosomes and translocated to the cytoplasm to begin the classical MHC class I antigen presentation pathway. These findings suggest that the autophagic machinery in HSV-1-infected macrophages contributes to the “cross-presentation” of endogenous viral antigen via the vacuolar pathway, supporting the current notion that antigen presenting cells (APCs) divert classical antigen presentation routes destined specifically for MHC class I or II, allowing substantial cross-talk among endomembrane organelles in order to maximize immune response efficiency against both endogenous and exogenous microbial antigens [42]. Indeed, a potential link between autophagy and antigen cross-presentation has emerged from the observation that silencing autophagy genes such as Beclin1 or Atg12 in antigen-donating tumor cells caused a decrease in cross-presentation [43]. In conclusion, host cells hire the autophagic machinery to participate in both intrinsic and adaptive immune responses against HSV-1 replication and pathogenesis; autophagy fights against HSV-1 by xenophagic degradation of viral components and the delivery of viral antigen to MHC class I molecules.
MHC class II antigens were initially believed to originate from lysosomal proteolysis of microbial material endocytosed or phagocytosed from extracellular regions. However, up to 20% of natural ligands eluted from affinity-purified MHC class II molecules turned out to be derived from endogenous proteins, and autophagy has been implicated in their generation [44]. Particularly, starvation-induced autophagy in human B-lymphoblastoid cells profoundly altered the class II peptide repertoire by increasing MHC class II presentation of intracellular, rather than membrane or secreted, proteins. Indeed, autophagy was found to enable intracellular viral proteins access to the lysosome for degradation and subsequent MHC class II loading. For instance, endogenous Epstein–Barr virus nuclear antigen 1 (EBNA1), the dominant CD4+ T cell antigen of latent EBV infection, was shown to accumulate in lysosomes and autophagosomes when EBV-transformed lymphoblastoid cells (LCLs) were incubated with lysosomal acidification inhibitors [45]. In addition, suppression of autophagy diminished MHC class II-restricted CD4+ T cell recognition of EBNA1, further supporting the notion that EBNA1 antigen follows an autophagic route to lysosomes. The possibility of MHC class II processing of exogenous EBNA1 can be excluded because of the study demonstrating that LCLs were incapable of sensitizing bystander B cells for EBNA1-specific CD4+ T cell recognition [46]. Collectively, these observations suggest that autophagy in EBV-transformed B cells promotes MHC class II processing of endogenous EBNA1 for CD4+ T cell recognition, implicating autophagy in viral antigen cross-presentation. A further study showed that autophagosomes are constitutively fused with MHC class II-loading compartments in professional APCs, such as B cells, monocytes, and dendritic cells, as well as in epithelial cell lines with IFN-γ-inducible MHC class II expression [47]. Notably, targeting viral antigens for autophagic degradation facilitated CD4+ T cell recognition. Several types of target cells (HeCat epithelial cells, B-LCLs, and DCs) expressing influenza A virus matrix protein 1 (M1) fused with autophagosomal marker LC3 displayed up to a 20-fold increase of MHC class II presentation to M1-specific CD4+ T cell clones compared to cells expressing M1 alone. However, M1-specific CD8+ T cell recognition remained unaffected, suggesting that the steady-state level of autophagy contributes to host adaptive immune responses against influenza virus by constitutively delivering M1 for MHC class II presentation and subsequent CD4+ T cell stimulation. In summary, autophagy is found to have an unprecedented function in facilitating the processing and presentation of various viral antigens derived from HSV-1, EBV, and influenza A virus. Autophagy may therefore be a novel target for vaccine development that allows specific boosting of host adaptive immune responses against viral infections.
Subversion of autophagy by viruses
Since autophagy is one of the emerging antiviral sentinels employed by the host immune system, certain viruses have successfully evolved to develop immune evasion programs that thwart this cell-autonomous defense response for their benefit. The herpesviruses may be one of the best examples of viruses that have co-evolved with their hosts, establishing a life-long latency after the primary lytic infection is cleared. As part of their strategy to compromise host immune surveillance, all three subfamilies of herpesviruses (α-, β-, and γ-herpesvirus) encode viral proteins that undermine the autophagy pathway, since it would otherwise participate in hindering viral replication and pathogenesis.
α- and β-herpesvirus proteins
As discussed above, the α-herpesvirus HSV-1 ICP34.5 inhibits the induction of autophagy by targeting the central modulator of autophagy, Beclin1, and one of its upstream signaling cues, PKR-mediated phosphorylation of eIF2α. This action is further reprised by US11, another HSV-1 protein, which also associates with PKR and hinders eIF2α phosphorylation, ensuring HSV-1-mediated blockade of host autophagic responses [48]. Moreover, HSV-1-encoded glycoprotein B (gB) binds to PERK and obstructs the phosphorylation of PERK and its substrate eIF2α [49]. Collectively, these three viral proteins potently inhibit eIF2α phosphorylation as part of the anti-autophagic strategy of HSV-1. Indeed, this may not be too surprising given that a robust synthesis of viral proteins during the productive stage of an infection should overwhelm the protein folding capacity of the endoplasmic reticulum (ER) to the point of triggering the unfolded protein response (UPR) in host cells to relieve ER stress. UPR and autophagy may be mechanistically linked as one arm of UPR leads to phosphorylation of eIF2α, serving to attenuate global protein translation [50]. In fact, ER stress has been shown to induce autophagy in yeast [51, 52]. Thus, a complete inhibition of eIF2α phosphorylation would probably be one of the most efficient tactics used by viruses to concomitantly confer resistance to ER stress-induced autophagic degradation as well as UPR-mediated translational shutoff. Analogous to α-herpesviruses, human cytomegalovirus (HCMV) belonging to the β-herpesvirus subgroup can potently counteract cellular autophagy responses even though UPR is induced, as evidenced by the accrual of phosphorylated eIF2α, its downstream transcription factor ATF4, and the spliced form of the XBP-1 transcript [53]. Notably, HCMV has been shown to drastically inhibit autophagosome formation in primary human fibroblasts [54]. Albeit not yet completely understood, one possible mechanism by which HCMV antagonizes host autophagy responses is via the stimulation of the mTOR signaling pathway as HCMV infection activates this pathway under normal conditions [54, 55], but treatment of rapamycin, a potent autophagy-inducing agent that inhibits mTOR, is incapable of relieving HCMV-mediated autophagy suppression. However, it should also be noted that HCMV-infected cells remained resistant to autophagy induction when treated with an mTOR-independent autophagy activator lithium chloride, indicating that HCMV can counteract host autophagy responses via both mTOR-dependent and -independent pathways.
γ-herpesvirus viral Bcl-2
In order to antagonize autophagy, γ-herpesvirus also have evolutionarily acquired viral homologues of B cell lymphoma-2 (Bcl-2), including BHRF1 and BALF-1 of EBV, orf16 of Kaposi’s sarcoma-associated herpesvirus (KSHV) and herpesvirus saimiri (HVS), and M11 of murine γ-herpesvirus 68 (γ-HV68) [56]. Early studies on viral Bcl-2 (vBcl-2) mainly explored their roles in abrogating cellular apoptotic responses. Expressed during its early lytic phase, vBcl-2 of KSHV (orf16) efficiently neutralizes Bax toxicity in yeast and suppresses apoptosis induced by Sindbis virus infections or by the over-expression of Bax or viral cyclin (vCyclin) in cultured cells [57–59]. Similarly, vBcl-2 of γ-HV68 (M11) has been implicated to prevent in vitro apoptotic cell death induced by a wide array of stimuli such as Fas, TNF-α, dexamethasone, γ-irradiation, and CD3ε ligation by targeting proapoptotic Bcl-2 family proteins Bax and Bak, which would otherwise promote the release of cytochrome c from the mitochondria [60, 61]. Beyond its established anti-apoptotic functions, the ability of cellular Bcl-2 to suppress autophagy has surfaced as a novel Bcl-2 function in the past few years. Accordingly, KSHV and γ-HV68 viral Bcl-2 proteins have been shown to mimic their cellular counterpart and attenuate autophagy through a direct interaction with Beclin1 [9, 62]. Indeed, KSHV-encoded vBcl-2 has been found to dampen Beclin1-mediated autophagy more potently than cellular Bcl-2 [63]. Furthermore, structural and biochemical analyses revealed that, compared to cellular Bcl-2, γ-HV68 M11 associates with Beclin1 with a markedly higher affinity and inhibits autophagosome formation more efficiently [64]. In conjunction with these findings, it has been demonstrated that while cellular Bcl-2 contains a regulatory loop that is phosphorylated by JNK1 to allow it to dissociate from Beclin1 and activate autophagy during starvation, vBcl-2 lacks this phosphorylation site-containing regulatory loop and thus continuously associate with Beclin1 [65]. These observations collectively indicate that vBcl-2 has evolved to exhibit an enhanced capacity for autophagy inhibition compared with its cellular counterpart.
Structural analyses also showed that Beclin1 possesses a putative BH3 domain that allows docking into the hydrophobic surface groove of both cellular and viral Bcl-2. Indeed, the hydrophobic surface groove also serves as a binding pocket for Bax and Bak, indicating that the dual functions of Bcl-2 in antagonizing apoptosis and autophagy involve the same structural cassette. In fact, the binding affinity of γ-HV68 M11 with Beclin1 was significantly higher than Bax/Bak or the upstream eight BH3-only proteins (Bad, Bik, Bim, Bid, Bmf, Puma, Noxa, and Hrk) [64], suggesting that vBcl-2 predominantly targets Beclin1 to robustly inhibit autophagy rather than apoptosis. A recent mutational study has led to the genetic and functional separation of the anti-apoptotic and anti-autophagic activities of M11: the Δα1 mutant that was impaired in anti-autophagy activity retained anti-apoptotic activity, while the ΔBH2 mutant behaved in an opposite fashion [66]. These observations indicate that despite their structural overlap, Beclin1 and pro-apoptotic Bcl-2 family proteins associate with vBcl-2 through two discrete modes of binding that are dependent on the distinct sequences of vBcl-2. Somewhat paradoxically, the anti-autophagic and anti-apoptotic activities of M11 are critical for the chronic infection, rather than the acute infection, of γ-HV68 despite the fact that vBcl-2 is primarily expressed during its lytic cycle. Strikingly, a M11 recombinant mutant virus defective in anti-apoptosis activity was largely compromised in ex vivo reactivation frequency, while a mutant virus lacking anti-autophagy activity was impaired in the maintenance of latency without affecting the initial establishment of latency. However, both M11 mutant viruses replicated as efficiently as wild-type during the acute phase of infection in the lungs of mice, and no evident differences were observed between wild-type and mutant viruses in their ability to establish a latent infection in the mouse spleen. These observations collectively demonstrate the discrete roles of M11 in the maintenance of viral latency and the subsequent ex vivo reactivation depend on evading two distinct host defense programs, autophagy and apoptosis, respectively. In support of this, the vBcl-2 transcript was previously detected in latently infected tissues [67], further confirming the role of vBcl-2 in the maintenance of viral persistency via the evasion of autophagy-mediated host innate immunity in vivo. Nonetheless, further studies are necessary to address why and how autophagy dismantles chronic viral infection. At present, several mechanisms can be envisioned: (1) Induction of xenophagy may promote degradation of viral proteins required for the maintenance of latency as observed with Sindbis virus and herpes simplex virus-1. (2) Beclin1-mediated autophagy-associated cell death may be responsible for elimination of latently infected cells as a Beclin1 mutant lacking the Bcl-2-binding ability has been shown to trigger caspase-independent cell death through autophagy [63]. Despite the consensus that autophagy is generally an adaptive response to sublethal stress for cytoprotection, this is one of the many new findings that has led to the recognition of autophagy as being involved in cellular suicide under certain circumstances, such as tumor suppression. Somewhat paradoxically, if autophagy destroys a substantial portion of the cytosol and organelles beyond a particular threshold, it can result in irreversible atrophy which ultimately leads to a cellular demise distinct from apoptosis or necrosis [68]. Although an increased number of autophagosomes has been detected in some dying cells, it is still unclear whether these autophagic vesicles genuinely contribute to cell death (autophagy-induced cell death or cell death by autophagy) or are merely a feature of a cell that has failed to salvage itself via self-cannibalization (autophagy-associated cell death or cell death with autophagy). However, the genetic ablation of essential autophagy genes expedites rather than attenuates cell death, emphasizing the predominant survival function of autophagy. Thus autophagy-associated cell death rather than autophagy-induced cell death is the favored model [24]. In any case, it seems reasonable to speculate that the up-regulation of autophagy followed by cell death may compromise the viral life cycle through the elimination of its hosts. (3) Given the fact that autophagy induction during metabolic stress has been shown to accompany cell cycle arrest [69], it is plausible to speculate that autophagy is involved in cell cycle arrest to hinder the proliferation of latently infected cells to restrict the replenishment of a persistent viral reservoir. In this case, vBcl-2 may exert dual functions in preventing autophagy as well as apoptosis to promote cell cycle progression during persistent infection and reactivation from latency, respectively. In support of this notion, KSHV vBcl-2 has been shown to obstruct apoptosis and promote cell cycle progression in concert with vCyclin [59]. (4) Based on previous studies demonstrating that type I and II IFNs are potent inhibitors of γ-HV68 reactivation from latency, it is tempting to hypothesize that autophagic trafficking mediates the delivery and loading of viral DNA to an as-of-yet undefined DNA sensing molecule to activate interferon signaling [70, 71]. In this scenario, interferon production facilitated by the autophagic machinery would ultimately disrupt viral persistency since a low level of spontaneous reactivation is necessary for maintaining a latent viral pool. (5) As exemplified by the EBV EBNA1, autophagy may participate in the processing and presentation of viral latent antigens to MHC molecules to trigger host adaptive immune responses. Further studies will be important to examine the discussed possibilities and delineate precise mechanism by which autophagy restricts the γ-herpesviral latency.
γ-herpesvirus viral FLIP
γ-herpesviruses also carry homologs of cellular FLICE-like inhibitor protein (cFLIP) to suppress the host autophagic machinery. As seen with Bcl-2, early studies primarily characterized cFLIP and viral FLIPs (vFLIPs) as inhibitors of death receptor-mediated apoptosis [72, 73]. The death effector domains (DEDs) of both cFLIP and vFLIP have been shown to directly compete with pro-caspase-8 for the interaction with adaptor proteins including Fas-associated via death domain (FADD) or TNFR-associated via death domain (TRADD) for binding to their respective death receptors, Fas and TNF, thereby preventing the assembly of the death-induced signaling complexes (DISC). KSHV-encoded vFLIP has also emerged as a potent activator of the NF-κB pathway by stimulating TNFR-assoicated factor 2 (TRAF2) and its downstream signaling modulators, the IKKαβγ complexes. Beyond this, a recent study has revealed that herpesviral FLIPs (KSHV, HVS, and MCV) as well as the long and short forms of cFLIP are capable of antagonizing autophagy induced by nutrient starvation or rapamycin treatment [74]. Mutagenesis analyses show that the anti-autophagic function of KSHV vFLIP is genetically separable from its anti-apoptotic and NF-κB-activating functions. Surprisingly, the E2-like Atg3 of the LC3/Atg8 conjugation system was identified to be a direct interaction partner of vFLIP and cFLIP. Indeed, ectopic expression of vFLIP resulted in a significant reduction of the interaction affinity between endogenous Atg3 and LC3, indicating that vFLIP inhibits autophagy by preventing Atg3 from binding and processing LC3 during autophagosome elongation. Moreover, ectopic expression of KSHV vFLIP was further shown to effectively repress autophagy-associated cell death induced by rapamycin, also known as a potent anti-tumor and immunosuppressive drug against KSHV-related diseases such as KSHV-induced Kaposi’s sarcoma (KS) and primary effusion lymphoma (PEL) [75, 76]. Conversely, ectopic expression of a vFLIP mutant lacking the Atg3 binding ability displayed minimal or no effect on rapamycin-induced growth inhibition and autophagy-associated cell death. These observations collectively indicate that the vFLIP-Atg3 interaction counteracts rapamycin-induced growth inhibition and autophagy-associated cell death. Remarkably, the ten amino acids of DED1 α2 helix and twelve amino acids of DED2 α4 helix of KSHV vFLIP are individually sufficient for Atg3 binding. Furthermore, the vFLIP-derived peptides effectively target the FLIP itself, resulting in the elevation of Atg3-LC3 interaction as well as the augmentation of autophagy. Consequently, the FLIP peptides demonstrate a potent in vitro and in vivo autophagy-associated cell death activity toward tumor cells. In conclusion, γ-herpesviruses seem to have acquired viral versions of Bcl-2 and FLIP in order to circumvent host apoptosis- and autophagy-mediated immune responses, thereby allowing them to permanently reside in their hosts as obligatory intracellular parasites. Given the frequent association between the onset of virus-induced oncogenesis and life-long persistent infection, it is also plausible to speculate that γ-herpesviral Bcl-2 and FLIP may contribute to pathogenesis partly by undermining the tumor suppressive function of the host autophagic machinery.
Human immunodeficiency virus-1 Env and Nef
In addition to herpesviruses, human immunodeficiency virus-1 (HIV-1), a single-stranded RNA virus in the genus of lentivirus of Retroviridae family, is another virus notorious for its ability to escape and thwart the host immune system. As the causative agent of acquired immunodeficiency syndrome (AIDS), HIV-1 induces an array of alterations in the host cell, including the autophagic machinery, to modulate the intracellular milieu in favor of viral survival and propagation. Studies on the relationship between HIV-1 infection and autophagy are still primitive. Yet, commensurate with its reputation as a sophisticated virus, HIV-1 seems to communicate with the host autophagic machinery in a highly complex manner. As will be discussed in the following section, autophagy, triggered by a fusogenic function of HIV-1 envelope glycoprotein (Env), has been implicated as a prerequisite for apoptotic cell death of uninfected bystander CD4+ T cells, thus potentially contributing to the depletion of the CD4+ T cell population and immunodeficiency [77–79]. However, there have been reports demonstrating that autophagy is suppressed in myeloid, lymphoid, and neuronal cells upon HIV-1 infection. Monocytes or CD4+ T cells infected with HIV-1 were shown to down-regulate the formation of autophagic vacuoles and the expression of proteins involved in autophagy such as LC3-PE and Beclin1 [80]. In agreement with this finding, autophagy was repressed in CD4+ T cells productively infected with either the X4 or R5 strain of HIV-1 [79], suggesting that HIV-1 is capable of antagonizing Env-mediated apoptosis in infected CD4+ T cells by suppressing autophagy, thereby allowing viral replication to occur. Moreover, an HIV-1 accessory protein called negative factor (Nef) has been described to block autophagosomal maturation [81]. While Nef expression in macrophages did not induce autophagic proteolysis of long-lived proteins, it resulted in the accumulation of autophagosomes and LC3-PE, the latter of which remained unaffected after treatment of bafilomycin A1. Accordingly, Nef expression prevented autophagic degradation of HIV-1 biosynthetic intermediates or virions. Nef likely does this by targeting the C3-PI3K complex as Nef associates with Beclin1, altering the subcellular distribution of Vps34. These findings collectively indicate that Nef blocks autophagic flux, especially the late stage of autophagy, which could otherwise potentially impede production of viral progenies through xenophagy. In line with these observations, a recent study has found that HIV-1 evades early immune responses by rapidly shutting down autophagy in dendritic cells (DCs) [82]. The inhibitory effect of HIV-1 on autophagy was dependent on the interaction between HIV-1 Env and DC surface receptors, most likely CD4, leading to the activation of the mTOR signaling pathway. Moreover, silencing the expression of autophagy genes in DCs resulted in the enhancement of cell-associated HIV-1 and viral transfer from DCs to CD4+ T cells. Surprisingly, autophagy-deficient DCs displayed changes in both TLR4- and TLR8-mediated responses and in the inhibition of MHC-II-dependent HIV-1 antigen presentation, indicating that HIV-1-mediated suppression of autophagy leads to impaired innate and adaptive immune response of DCs. Thus, this provides, at least in part, an explanation of how a substantial fraction of HIV-1 can evade early immune responses by DCs in the mucosal surface and be transmitted to CD4+ T cells. It is also worth noting that the ability of HIV to suppress autophagy may contribute to HIV-induced neurological disorders such as HIV-associated dementia and HIV-induced encephalopathies, as disturbances of neuronal homeostasis arising from accumulated protein aggregates have been implicated in their pathogeneses. Based on the accumulated evidence suggesting a direct correlation between neuronal autophagy defects and neurodeneration, it will be thus, important to elaborate the molecular mechanism by which HIV inhibits autophagy to establish neural pathogenesis [83].
Influenza virus M2
A recent study has added influenza A virus, a segmented negative-sense single-stranded RNA virus that is a epidemic and pandemic threat to the human population, to the growing list of viruses that interfere with the cellular autophagy pathway. It was demonstrated that A549 human lung epithelial cells infected with various strains of influenza A virus (A/Aichi/68 (H3N2), A/WSN/33 (H1N1), and A/PR8/34 (H1N1)) displayed an accumulation of autophagosomes, resulting in the generation of immobile large perinuclear autophagosomes [84]. This phenotype was further confirmed to be the consequence of a blockade in autophagosomal fusion with lysosome, and not a general enhancement of autophagosome generation. Additionally, influenza A virus matrix protein 2 (M2), a proton-selective ion channel that plays a pivotal role in influenza A virus assembly and budding [85], was identified to be necessary and sufficient for the inhibition of autophagosomal maturation in a manner that is independent of its ion channel activity. Expression of M2 recapitulated the blockade of autophagosomal degradation observed after influenza A virus infection. Analogous to HIV-1 Nef, influenza A virus M2 associated with Beclin1, but it is not yet clear if this interaction indeed debilitates the function of the Beclin1-containing complex in autophagosomal maturation, necessitating further investigations. Interestingly, autophagy-deficient cells exhibited no difference in the release of viral progenies compared with autophagy-competent cells, indicating that viral replication does not require autophagosomes nor does autophagy attenuate viral replication. Rather, the inhibition of autophagy by M2 appears to compromise the survival of influenza A virus-infected cells, as Atg5-deficient MEFs exhibited a notably elevated susceptibility to apoptosis upon influenza A virus infection. Conversely, allowing autophagy to progress through deletion of M2 diminished influenza A virus-induced apoptosis. Overall, these observations suggest that influenza A virus induces apoptosis by compromising autophagy, one of the major prosurvival pathways of the host cell. Influenza A virus seemingly strives to modulate the delicate trade-off balance between apoptosis and autophagy, as appropriately timed apoptosis at moderate levels appears crucial for its replication. However, the functional advantages of augmented host cell death during viral infection remain largely unexplored. Also, further investigations are necessary to delineate how and why Influenza A virus M2 suppresses autophagosomal maturation. Based on the role of autophagy in the cross-presentation of M1 antigens as discussed in the previous section, a reasonable conjecture may be that M2 inhibits autophagosomal fusion with lysosome in order to antagonize autophagy-mediated MHC class II presentation to the M2-specific CD4+ T cell clones. Somewhat contradictory to the above report, a recent study has shown that the A/WSN/33 (H1N1) and A/chicken/Beijing/04 (H9N2) strains of influenza A virus are capable of enhancing autophagosome formation to ameliorate viral replication [86]. Furthermore, inhibition of autophagy via pharmacological reagents (3-methylademine or wortmannin) or RNA interference-mediated suppression of Beclin1 or LC3 impaired viral yield in A549 cells as opposed to Atg5-deficient MEFs. This suggests that the modulation of autophagy may be cell-type dependent, necessitating further elaborate investigations.
Beyond sabotage: virus-induced autophagy
An astonishing number of viruses appear to go beyond evasion or subversion of host autophagy pathway. Instead, they aggressively trigger (certain stages of) autophagy or exploit the cellular autophagic machinery to benefit their replication. For instance, all plus-stranded RNA viruses have been implicated to elicit the remodeling of cellular membranes of unknown origin to take advantage of its surface as a site of viral genome replication [87], and autophagic vacuoles may be one of the targets for certain viruses. Two members of the picornavirus family, poliovirus and coxsackievirus B3 (CVB3), have been shown to activate the formation of cytoplasmic vesicles resembling autophagosomes during the course of infection [88, 89]. In both studies, virus infection induced an increased number of double-membrane vesicles accompanied by hallmarks of cellular autophagosomes, including colocalization of LC3 and LAMP1, increased LC3-II/LC3-I ratio, and accumulation of GFP-LC3 puncta. Also, pharmacological or genetic inhibition of autophagosome formation dampened viral replication, while induction of autophagy by rapamycin or starvation fostered viral replication. Moreover, the endogenous level of p62 protein remained unaltered after CVB3 infection, suggesting that viral infection induces autophagosome formation without promoting lysosome-mediated protein degradation. Hence, autophagosomes appear to simply supply the membranous support for the formation of a viral RNA replication complex. At present, however, the underlying molecular mechanism remains to be further determined. In the case of poliovirus, viral proteins 2BC and 3A were shown to be necessary for the formation of autophagic vacuoles and the lipidation of LC3. Yet, it is not clear if both viruses actively induce formation of autophagosome-like structures and/or if they block the maturation of formerly formed autophagosomes. It is also possible that autophagosome-like membranes assist the release of viral progeny from infected cells without cell lysis. Silencing protein expression of LC3 or Atg12 decreased the amount of viral particles in the extracellular milieu more than intracellular virions, suggesting that poliovirus hijacks the autophagic vesicle to utilize it as a membrane scaffold for viral replication and, ultimately, non-lytic egress. However, the contribution of autophagy induction to in vivo pathogenesis of poliovirus and CVB3 has not yet been addressed.
Another plus-stranded RNA virus, hepatitis C virus (HCV), has been shown to induce the early, non-degradative stage of autophagy via stimulation of ER stress [90]. All genotypes of HCV, 1a, 1b, and 2a, were able to trigger the accumulation of autophagosomes but not autolysosomes [91]. Analogous to CVB3, HCV did not facilitate autophagic degradation of long-lived proteins, indicating that HCV infection induces the accumulation of autophagosomes by suppressing their fusion with lysosomes. Indeed, HCV induced an incomplete autophagic response by introducing a persistent induction of ER stress and the subsequent UPR via three different sensors—PERK, ATF6, and IRE1. Furthermore, HCV-induced autophagosomes were shown to promote viral replication, as the alleviation of ER stress and the siRNA-mediated suppression of UPR sensors or autophagy proteins all resulted in repression of HCV RNA replication. Indeed, the autophagic machinery was required for the translation of incoming viral RNA, but not for entry or egress. Besides this, autophagy proteins (Beclin1, Atg4B, Atg5, and Atg12) were required to initiate viral RNA translation but not to maintain HCV replication once infection was established [92]. These observations indicate that the host factors required for the translation of incoming HCV RNA are distinct from those required for the translation of progeny viral RNA. Alternatively, the autophagic machinery may support the delivery of incoming viral RNA to the translation apparatus and/or the recruitment cellular factors required to initiate translation.
In contrast to its anti-autophagic activity discussed in the previous section, HIV-1 can also enhance autophagy. HIV-1 seems to cleverly utilize the autophagic machinery to trigger immunodeficiency through up-regulation of autophagy in CD4+ T cells. Although both infected and uninfected CD4+ T cells can perish during an HIV-1 infection, HIV-1-induced bystander CD4+ T cell death is increasingly being recognized to be central for immunodeficiency during the course of AIDS development. This is because the vast majority of CD4+ T cells undergoing apoptosis as well as the peripheral blood and lymph nodes of HIV patients have been found to be uninfected [93]. As mentioned in the previous section, X4 or R5 HIV-1-infected cells that express envelope glycoproteins (gp41 and gp120; Env) induced autophagy-mediated apoptotic cell death not in themselves but in neighboring bystander CD4+ T cells via fusion with a coreceptor CXCR4 or CCR5, respectively [77, 79]. However, this bystander effect was not observed in the case of macrophages where HIV-1 can infect and replicate as well. These observations may at least partly elucidate the mechanisms by which HIV-1 selectively kills uninfected CD4+ T cells while leaving the population of macrophages intact in infected patients. Interestingly, autophagy was rather induced in monocyte-derived macrophages infected with HIV-1, which was opposed to HIV-1-infected CD4+ T cells. Indeed, HIV-1 Gag protein was shown to induce the early stage of autophagosome formation through a direct interaction with LC3 in macrophages [81]. The association of Gag with autophagosomes augmented HIV-1 yields and processing of Gag, a critical step in the assembly and release of the virions. As HIV-1 replicates its genome in the nucleus, it is less likely that HIV-1 induces autophagy in order to utilize autophagosome-like structures in the cytoplasm as membrane scaffolds for the replication of their genomes. Instead, it is more reasonable to speculate that autophagic vacuoles may be involved in morphogenesis and egress of newly produced HIV-1 virions by, for instance, directing them to multi-vesicular bodies in macrophages [94]. These studies collectively suggest that virus-induced autophagy in macrophages and uninfected CD4+ T cells likely contributes, at least in part, to an enhancement of HIV-1 replication and to immunodeficiency, respectively. Moreover, a genome-wide siRNA screening in HeLa cell derivatives has found a number of new autophagy genes important in HIV-1 replication [95]. Interestingly, these newly identified genes—Atg7, Atg8, Atg12, and Atg16L2—are all components of the ubiquitin-like conjugation system involved in vesicle elongation, further suggesting that autophagosome formation is critical for productive HIV-1 replication. However, the in vivo role of these genes in HIV-1 infection in natural host cells should be further examined. Moreover, one cannot exclude the possibility that these genes possess alternative autophagy-independent functions that affect HIV-1 replication. The global role of autophagy in HIV-1 replication and pathogenesis yet remains perplexing since HIV-1 possesses two opposing abilities in modulating cellular autophagy in a cell type-specific manner.
Likewise, herpesviruses arry two sides of autophagy controlling activity, and this may may be due to the complicate life cycles. Unlike several herpesviral proteins that suppress autophagy as discussed above, an EBV-encoded latent membrane protein (LMP1) has been shown to modulate B cell physiology by inducing autophagy [96]. The intermediate levels of LMP1 induce proliferation, while its high levels trigger inhibition of general protein synthesis and cytostasis. Thus, EBV cunningly exploits the cellular autophagic machinery in order to control LMP1 protein level. Four out of six transmembrane domains of LMP1 were responsible for inducing autophagy in a dose-dependent manner. Furthermore, inhibition of autophagy via knockdown of Beclin1 or Atg7 in EBV-infected cells resulted in an accumulation of LMP1 and a reduced ability to form colonies, indicating that EBV LMP1 induces autophagic degradation to limit its accumulation and regulate host cell behavior. However, the molecular mechanism of LMP1-mediated induction of autophagy remains uncharacterized. One possibility would be the phosphorylation of eIF2α, as high levels of LMP1 have been shown to induce general protein synthesis by activating PERK, triggering eIF2α phosphorylation [97]. Additionally, a neurotropic α-herpesvirus VZV has also been shown to induce autophagy in cultured cells and human zoster vesicles but not in normal skin [98]. Finally, in contrast to the anti-autophagic roles of KSHV-encoded vBcl-2 and vFLIP, replication and transcription activator (RTA), an essential viral protein for KSHV lytic reactivation, has been recently described to induce autophagy to facilitate lytic replication [99]. This study demonstrated that inhibition of autophagy suppressed RTA-mediated lytic gene expression and KSHV lytic replication, while an enhanced level of autophagy was detected during lytic reactivation from latency. Moreover, over-expression of RTA resulted in increased numbers of both autophagosomes and autolysosomes, and enhancement of GFP-LC3 puncta staining, collectively suggesting that RTA possesses the ability to activate autophagy. Yet, it is not clear how RTA, a typical transcription factor comprised of a DNA-binding domain and an activation domain, can affect autophagy, which occurs in the cytoplasm. Taken together, it seems unclear yet whether EBV, VZV, and KSHV indeed actively induce autophagy to enhance their own replication or if the elevated level of autophagy stems from the attempt of host to combat these viruses.
Conclusion
Autophagy is an evolutionarily conserved unique membrane trafficking event that allows self-cannibalism to control the quality and quantity of cytoplasmic entities for cellular homeostasis. Recent advances in autophagy research have unveiled the previously overlooked importance of autophagy in various aspects of human pathophysiology including cell-autonomous defense against intruding microbes. In this review, we have highlighted recent studies, presenting a complex picture of the diverse roles of autophagy in virus life cycle and pathogenesis.
Converging evidence suggests that the autophagy pathway and/or autophagy genes play integral roles in intrinsic, innate, and adaptive immune responses against viral infection and pathogenesis. Intrinsic xenophagy has been shown to alleviate neuropathogenesis of SIN and HSV-1 infection by sequestering virions or viral components for lysosomal degradation. A series of studies have also observed that the autophagy pathway and/or autophagy genes play pivotal roles in homeostatic regulation of innate immune responses against VSV by negatively regulating RLR signaling, while activating TLR signaling in different cell types. Furthermore, autophagy is found to have a novel function in adaptive immunity against HSV-1, EBV, and influenza A virus as a facilitator of viral antigen processing and presentation for T cell recognition. However, certain viruses including HSV-1, HCMV, KSHV, HVS, MCV, and γ-HV68 have developed mechanisms to evade and, moreover, antagonize autophagy-associated host antiviral defense responses. Notably, γ-herpesviruses have evolved to escape autophagy-mediated immune programs, thereby contributing to the maintenance of latent infection and virus-induced oncogenesis. HIV-1 infection also suppresses autophagy to evade early immune responses in DCs and create an environment favorable for viral replication in CD4+ T cells. Additionally, HIV-1, influenza A virus, and HCV impair autophagic flux, especially the late stage of autophagy, which leads to the accumulation of autophagosomes. In contrast, given that the intracellular milieu provides obligate intracellular parasites with the opportunity to expropriate cellular machineries for their metabolic purposes [100], it is not surprising that certain viruses enhances autophagy for their benefit. Several RNA viruses such as Poliovirus, CVB3, and HCV, and HIV-1 induce the formation of double-membrane vesicles resembling autophagosomes to foster viral replication or non-lytic egress. Moreover, HIV-1-induced autophagy has been proposed to partly contribute to immunodeficiency by selectively triggering apoptotic cell death of uninfected bystander CD4+ T cells and thus depletion of the T cell population. Finally, certain DNA viruses such as EBV, KSHV, VZV, and HBV are also capable of promoting autophagy to benefit their replication and/or pathogenesis. Despite these extensive studies, however, our understanding of the relationship between autophagy and virus is yet in its infancy, partly because the role of host autophagy is often virus- and/or cell type-specific. For example, deletion of autophagy genes has exhibited differential effects in different cell populations. Thus, intensive research aimed at distinguishing the roles of autophagy proteins in the classical autophagy pathway versus as-of-yet undetermined autophagy-independent processes may help delineate how these homeostatic checkpoint proteins have functionally diversified to antagonize and/or aid viral replication and pathogenesis. The insights obtained from these studies may provide therapeutic strategies and, eventually, opportunities for the discovery of novel drug targets of viral infection and its complications.
References
Klionsky DJ (2007) Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 8:931–937
Levine B, Kroemer G (2008) Autophagy in the pathogenesis of disease. Cell 132:27–42
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N (2009) Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20:1981–1991
Yan Y, Flinn RJ, Wu H, Schnur RS, Backer JM (2009) hVps15, but not Ca2+/CaM, is required for the activity and regulation of hVps34 in mammalian cells. Biochem J 417:747–755
Lindmo K, Stenmark H (2006) Regulation of membrane traffic by phosphoinositide 3-kinases. J Cell Sci 119:605–614
Suzuki K, Kubota Y, Sekito T, Ohsumi Y (2007) Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12:209–218
Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T (2009) Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396
Sun Q, Fan W, Chen K, Ding X, Chen S, Zhong Q (2008) Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 105:19211–19216
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU (2006) Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 8:688–699
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, Pledger WJ, Wang HG (2007) Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 9:1142–1151
Cecconi F, Di Bartolomeo S, Nardacci R, Fuoco C, Corazzari M, Giunta L, Romagnoli A, Stoykova A, Chowdhury K, Fimia GM, Piacentini M (2007) A novel role for autophagy in neurodevelopment. Autophagy 3:506–508
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402:672–676
Noda T, Fujita N, Yoshimori T (2008) The Ubi brothers reunited. Autophagy 4:540–541
Nakatogawa H, Ichimura Y, Ohsumi Y (2007) Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 130:165–178
Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–658
Klionsky DJ (2005) The molecular machinery of autophagy: unanswered questions. J Cell Sci 118:7–18
Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL (2004) Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 117:4837–4848
Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi Y, Yoshimori T (2002) SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Struct Funct 27:29–37
Tamai K, Tanaka N, Nara A, Yamamoto A, Nakagawa I, Yoshimori T, Ueno Y, Shimosegawa T, Sugamura K (2007) Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem Biophys Res Commun 360:721–727
Surpin M, Zheng H, Morita MT, Saito C, Avila E, Blakeslee JJ, Bandyopadhyay A, Kovaleva V, Carter D, Murphy A, Tasaka M, Raikhel N (2003) The VTI family of SNARE proteins is necessary for plant viability and mediates different protein transport pathways. Plant Cell 15:2885–2899
Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, Jung JU (2008) Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nat Cell Biol 10:776–787
Levine B (2005) Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120:159–162
Virgin HW, Levine B (2009) Autophagy genes in immunity. Nat Immunol 10:461–470
Kroemer G, Levine B (2008) Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 9:1004–1010
Bieniasz PD (2004) Intrinsic immunity: a front-line defense against viral attack. Nat Immunol 5:1109–1115
Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B (1998) Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 72:8586–8596
Orvedahl A, Alexander D, Talloczy Z, Sun Q, Wei Y, Zhang W, Burns D, Leib DA, Levine B (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1:23–35
Djavaheri-Mergny M, Maiuri MC, Kroemer G (2010) Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 29:1717–1719
Talloczy Z, Jiang W, HWt V, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci USA 99:190–195
Talloczy Z, HWt V, Levine B (2006) PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2:24–29
Markovitz NS, Baunoch D, Roizman B (1997) The range and distribution of murine central nervous system cells infected with the gamma(1)34.5- mutant of herpes simplex virus 1. J Virol 71:5560–5569
Mohr I, Sternberg D, Ward S, Leib D, Mulvey M, Gluzman Y (2001) A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J Virol 75:5189–5196
Soosaar JL, Burch-Smith TM, Dinesh-Kumar SP (2005) Mechanisms of plant resistance to viruses. Nat Rev Microbiol 3:789–798
Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP (2005) Autophagy regulates programmed cell death during the plant innate immune response. Cell 121:567–577
Shelly S, Lukinova N, Bambina S, Berman A, Cherry S (2009) Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30:588–598
Georgel P, Jiang Z, Kunz S, Janssen E, Mols J, Hoebe K, Bahram S, Oldstone MB, Beutler B (2007) Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology 362:304–313
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27:135–144
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A (2007) Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315:1398–1401
Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K (2007) The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci USA 104:14050–14055
Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A (2009) Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci USA 106:2770–2775
English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippe R, Desjardins M (2009) Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat Immunol 10:480–487
English L, Chemali M, Desjardins M (2009) Nuclear membrane-derived autophagy, a novel process that participates in the presentation of endogenous viral antigens during HSV-1 infection. Autophagy 5:1026–1029
Li Y, Wang LX, Yang G, Hao F, Urba WJ, Hu HM (2008) Efficient cross-presentation depends on autophagy in tumor cells. Cancer Res 68:6889–6895
Dengjel J, Schoor O, Fischer R, Reich M, Kraus M, Muller M, Kreymborg K, Altenberend F, Brandenburg J, Kalbacher H, Brock R, Driessen C, Rammensee HG, Stevanovic S (2005) Autophagy promotes MHC class II presentation of peptides from intracellular source proteins. Proc Natl Acad Sci USA 102:7922–7927
Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, Tuschl T, Munz C (2005) Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307:593–596
Munz C, Bickham KL, Subklewe M, Tsang ML, Chahroudi A, Kurilla MG, Zhang D, O'Donnell M, Steinman RM (2000) Human CD4(+) T lymphocytes consistently respond to the latent Epstein–Barr virus nuclear antigen EBNA1. J Exp Med 191:1649–1660
Schmid D, Pypaert M, Munz C (2007) Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 26:79–92
Mulvey M, Poppers J, Sternberg D, Mohr I (2003) Regulation of eIF2alpha phosphorylation by different functions that act during discrete phases in the herpes simplex virus type 1 life cycle. J Virol 77:10917–10928
Mulvey M, Arias C, Mohr I (2007) Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol 81:3377–3390
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274
Yorimitsu T, Nair U, Yang Z, Klionsky DJ (2006) Endoplasmic reticulum stress triggers autophagy. J Biol Chem 281:30299–30304
Bernales S, McDonald KL, Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4:e423
Isler JA, Skalet AH, Alwine JC (2005) Human cytomegalovirus infection activates and regulates the unfolded protein response. J Virol 79:6890–6899
Chaumorcel M, Souquere S, Pierron G, Codogno P, Esclatine A (2008) Human cytomegalovirus controls a new autophagy-dependent cellular antiviral defense mechanism. Autophagy 4:46–53
Kudchodkar SB, Yu Y, Maguire TG, Alwine JC (2004) Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol 78:11030–11039
Cuconati A, White E (2002) Viral homologs of BCL-2: role of apoptosis in the regulation of virus infection. Genes Dev 16:2465–2478
Cheng EH, Nicholas J, Bellows DS, Hayward GS, Guo HG, Reitz MS, Hardwick JM (1997) A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci USA 94:690–694
Sarid R, Sato T, Bohenzky RA, Russo JJ, Chang Y (1997) Kaposi’s sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat Med 3:293–298
Ojala PM, Tiainen M, Salven P, Veikkola T, Castanos-Velez E, Sarid R, Biberfeld P, Makela TP (1999) Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6. Cancer Res 59:4984–4989
Wang GH, Garvey TL, Cohen JI (1999) The murine gammaherpesvirus-68 M11 protein inhibits Fas- and TNF-induced apoptosis. J Gen Virol 80(Pt 10):2737–2740
Loh J, Huang Q, Petros AM, Nettesheim D, van Dyk LF, Labrada L, Speck SH, Levine B, Olejniczak ET, HWt V (2005) A surface groove essential for viral Bcl-2 function during chronic infection in vivo. PLoS Pathog 1:e10
Sinha S, Colbert CL, Becker N, Wei Y, Levine B (2008) Molecular basis of the regulation of Beclin 1-dependent autophagy by the gamma-herpesvirus 68 Bcl-2 homolog M11. Autophagy 4:989–997
Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122:927–939
Ku B, Woo JS, Liang C, Lee KH, Hong HS, Xiaofei E, Kim KS, Jung JU, Oh BH (2008) Structural and biochemical bases for the inhibition of autophagy and apoptosis by viral BCL-2 of murine gamma-herpesvirus 68. PLoS Pathog 4:e25
Wei Y, Pattingre S, Sinha S, Bassik M, Levine B (2008) JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30:678–688
Xiaofei E, Hwang S, Oh S, Lee JS, Jeong JH, Gwack Y, Kowalik TF, Sun R, Jung JU, Liang C (2009) Viral Bcl-2-mediated evasion of autophagy aids chronic infection of gammaherpesvirus 68. PLoS Pathog 5:e1000609
Roy DJ, Ebrahimi BC, Dutia BM, Nash AA, Stewart JP (2000) Murine gammaherpesvirus M11 gene product inhibits apoptosis and is expressed during virus persistence. Arch Virol 145:2411–2420
Maiuri MC, Zalckvar E, Kimchi A, Kroemer G (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol 8:741–752
Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM, Mills GB (2007) The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol 9:218–224
Barton ES, Lutzke ML, Rochford R, HWt V (2005) Alpha/beta interferons regulate murine gammaherpesvirus latent gene expression and reactivation from latency. J Virol 79:14149–14160
Steed AL, Barton ES, Tibbetts SA, Popkin DL, Lutzke ML, Rochford R, HWt V (2006) Gamma interferon blocks gammaherpesvirus reactivation from latency. J Virol 80:192–200
Djerbi M, Screpanti V, Catrina AI, Bogen B, Biberfeld P, Grandien A (1999) The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J Exp Med 190:1025–1032
Garvey TL, Bertin J, Siegel RM, Wang GH, Lenardo MJ, Cohen JI (2002) Binding of FADD and caspase-8 to molluscum contagiosum virus MC159 v-FLIP is not sufficient for its antiapoptotic function. J Virol 76:697–706
Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C, Jung JU (2009) FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol 11:1355–1362
Izzedine H, Wirden M, Launay-Vacher V (2005) Viral load and HIV-associated nephropathy. N Engl J Med 353:1072–1074
Sin SH, Roy D, Wang L, Staudt MR, Fakhari FD, Patel DD, Henry D, Harrington WJ Jr, Damania BA, Dittmer DP (2007) Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood 109:2165–2173
Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M (2006) Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 116:2161–2172
Denizot M, Varbanov M, Espert L, Robert-Hebmann V, Sagnier S, Garcia E, Curriu M, Mamoun R, Blanco J, Biard-Piechaczyk M (2008) HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy 4:998–1008
Espert L, Varbanov M, Robert-Hebmann V, Sagnier S, Robbins I, Sanchez F, Lafont V, Biard-Piechaczyk M (2009) Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoS ONE 4:e5787
Zhou D, Spector SA (2008) Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS 22:695–699
Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A, Federico M, Panganiban A, Vergne I, Deretic V (2009) Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. J Cell Biol 186:255–268
Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, Stalder R, Garcia E, Dinkins C, Leuba F, Wu L, Schwartz O, Deretic V, Piguet V (2010) Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity 32:654–669
Winslow AR, Rubinsztein DC (2008) Autophagy in neurodegeneration and development. Biochim Biophys Acta 1782:723–729
Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, Garcia-Sastre A, Munz C (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6:367–380
Chen BJ, Leser GP, Jackson D, Lamb RA (2008) The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol 82:10059–10070
Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J, Wang M, Gao GF (2009) Autophagy is involved in influenza A virus replication. Autophagy 5:321–328
Salonen A, Ahola T, Kaariainen L (2005) Viral RNA replication in association with cellular membranes. Curr Top Microbiol Immunol 285:139–173
Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 3:e156
Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H (2008) Autophagosome supports coxsackievirus B3 replication in host cells. J Virol 82:9143–9153
Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH (2008) Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061
Ait-Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R (2008) Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol 82:2241–2249
Dreux M, Gastaminza P, Wieland SF, Chisari FV (2009) The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci USA 106:14046–14051
Debatin KM, Fahrig-Faissner A, Enenkel-Stoodt S, Kreuz W, Benner A, Krammer PH (1994) High expression of APO-1 (CD95) on T lymphocytes from human immunodeficiency virus-1-infected children. Blood 83:3101–3103
Carter CA, Ehrlich LS (2008) Cell biology of HIV-1 infection of macrophages. Annu Rev Microbiol 62:425–443
Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ (2008) Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926
Lee DY, Sugden B (2008) The latent membrane protein 1 oncogene modifies B-cell physiology by regulating autophagy. Oncogene 27:2833–2842
Lee DY, Sugden B (2008) The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111:2280–2289
Takahashi MN, Jackson W, Laird DT, Culp TD, Grose C, Haynes JI 2nd, Benetti L (2009) Varicella-zoster virus infection induces autophagy in both cultured cells and human skin 83(11):5466–5476
Wen HJ, Yang Z, Zhou Y, Wood C (2010) Enhancement of autophagy during lytic replication by KSHV replication and transcription activator/RTA. J Virol 84(15):7448–7458
Bird PI, Trapani JA, Villadangos JA (2009) Endolysosomal proteases and their inhibitors in immunity. Nat Rev Immunol 9:871–882
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Autophagy.
Rights and permissions
About this article
Cite this article
Kim, H.J., Lee, S. & Jung, J.U. When autophagy meets viruses: a double-edged sword with functions in defense and offense. Semin Immunopathol 32, 323–341 (2010). https://doi.org/10.1007/s00281-010-0226-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-010-0226-8