
Abstract
Purpose
To characterize exposure–response relationships of AMG 386 in a phase 2 study in advanced ovarian cancer for the facilitation of dose selection in future studies.
Methods
A population pharmacokinetic model of AMG 386 (N = 141) was developed and applied in an exposure–response analysis using data from patients (N = 160) with recurrent ovarian cancer who received paclitaxel plus AMG 386 (3 or 10 mg/kg once weekly) or placebo. Reduction in the risk of progression or death with increasing exposure (steady-state area under the concentration-versus-time curve [AUCss]) was assessed using Cox regression analyses. Confounding factors were tested in multivariate analysis. Alternative AMG 386 doses were explored with Monte Carlo simulations using population pharmacokinetic and parametric survival models.
Results
There was a trend toward increased PFS with increased AUCss (hazard ratio [HR] for each one-unit increment in AUCss, 0.97; P = 0.097), suggesting that the maximum effect on prolonging PFS was not achieved at the highest dose tested (10 mg/kg). Among patients with AUCss ≥ 9.6 mg h/mL, PFS was 8.1 months versus 5.7 months for AUCss < 9.6 mg h/mL and 4.6 months for placebo. No relationship between AUCss and grade ≥3 adverse events was observed. Simulations predicted that AMG 386 15 mg/kg once weekly would result in an AUCss ≥ 9.6 mg h/mL in >90% of patients with median PFS of 8.2 months versus 5.0 months for placebo (HR [15 mg/kg vs. placebo], 0.56).
Conclusions
Increased exposure to AMG 386 was associated with improved clinical outcomes in recurrent ovarian cancer, supporting the evaluation of a higher dose in future studies.
Similar content being viewed by others

Introduction
Quantitative drug and disease modeling techniques are increasingly applied to today’s drug development process to inform critical decisions. By integrating complex data, the resulting deeper understanding of a drug’s action can guide go/no-go decision making, inform dose and study design selection, facilitate the development of combination therapies, and provide a better understanding of risk–benefit ratios [1, 2]. Use of such pharmacometric techniques may be particularly valuable in oncology, where phase 2 studies have historically been poor positive predictors of phase 3 outcomes [3, 4]. A key goal during early-phase development of novel cancer therapeutics is finding a dose that maximizes clinical benefit while maintaining an acceptable safety profile [5–7]. Because dose-ranging phase 2 studies are not frequently conducted in oncology [8], selecting an appropriate dose for a registrational study can present a considerable challenge. However, poor dose selection is one likely contributor to the historically limited ability of phase 2 studies to predict success of subsequent phase 3 trials [3, 4]. Population pharmacokinetic/pharmacodynamic modeling is an innovative tool that has shown promise when applied to the prediction of clinical endpoints [9, 10], identification of factors influencing clinical endpoints [11], and dose selection [12, 13]. Importantly, the US Food and Drug Administration (FDA) has suggested that exposure–response models may provide useful information for end-of-phase-2 dose-selection decisions [14]. Specifically, the FDA’s Critical Path Initiative [15] has identified quantitative modeling as a technique that may reduce uncertainty regarding dose selection.
AMG 386 (previously referred to as 2xCon4) is an investigational peptide-Fc fusion protein that mediates antiangiogenic effects by potently and selectively inhibiting the interaction of angiopoietin-1 and angiopoietin-2 with Tie2 [16]. Primary endpoint results from a phase 2 study of AMG 386 in combination with weekly paclitaxel for the treatment of recurrent ovarian cancer showed longer median progression-free survival (PFS) for patients receiving AMG 386 at 10 and 3 mg/kg once weekly (QW), compared with placebo (the data are described in the primary analysis [17]). Additional dose-exposure analyses suggested a dose–response effect across treatment arms.
Although the study strongly suggests that AMG 386 has antitumor activity in recurrent ovarian cancer, the dose–response effect raises the question of whether the tested doses maximized the risk–benefit ratio. The objective of the present analysis was to use population pharmacokinetic/pharmacodynamic models to explore whether doses of AMG 386 higher than those assessed in the phase 2 clinical study might result in further improvements in PFS while maintaining acceptable toxicity. Specifically, the models comprised a characterization of the pharmacokinetics of AMG 386 in recurrent ovarian cancer patients, an exposure–response analysis to assess the relationship between AMG 386 exposure and efficacy/toxicity endpoints, and exploration of potential clinical outcomes at doses higher than those evaluated in the phase 2 study.
Methods
Data sources
AMG 386 serum concentration-versus-time data for the population pharmacokinetic analysis were obtained from two clinical studies: a phase 1 first-in-human monotherapy study [18] in advanced solid tumors (n = 32) and a phase 2 study [17] in advanced ovarian cancer (n = 109). The phase 1 study was a sequential dose-escalation study of AMG 386 administered intravenously QW at 5 doses (0.3, 1, 3, 10, and 30 mg/kg) with 4–16 patients per dose group [18]. Intensive serum samples were collected at the end of infusion and at 2, 6, 24, 48, and 96 h after the infusion at weeks 1 and 4. Sparse pharmacokinetic (peak or trough) samples were collected before each weekly AMG 386 administration. The phase 2 study evaluated weekly paclitaxel plus AMG 386 at 10 mg/kg QW and 3 mg/kg QW, or placebo [17]. Pharmacokinetic samples were collected predose at weeks 1, 3, 5, and 9; every 8 weeks thereafter; and at the end of infusion at weeks 1 and 5. All patients provided written informed consent, and the study protocols were approved by an ethics committee at each participating center.
Efficacy and toxicity data for the exposure–response analysis were obtained from the phase 2 study [17]. The primary endpoint was PFS, defined as the time from randomization to the date of disease progression per Response Evaluation Criteria in Solid Tumors version 1.0 [19], clinical progression (per investigator), CA-125 progression (Gynecologic Cancer Intergroup criteria [20]), or death. Further study details are reported in the primary analysis [17].
Population pharmacokinetic analysis
A linear 2-compartment model was used to describe AMG 386 concentration data with population pharmacokinetic modeling using the nonlinear mixed-effect modeling software program NONMEM (version V, level 1.1; ICON Development Solutions, Ellicott City, MD) [21]. An exponential interindividual variability error term that assumed a log-normal distribution was included with all pharmacokinetic parameters (clearance [CL], distribution clearance [Q], and volumes of distribution for the central compartments [Vc] and peripheral compartments) in the model. Combined additive and proportional components were used to model residual intraindividual random error.
Five baseline variables (weight, sex, age, serum creatinine, and creatinine clearance [CrCL]) were tested with forward selection (P < 0.01) and backward elimination (P < 0.005) approaches for their effects on CL and Vc [22]. CrCL was calculated based on the Cockcroft-Gault formula [23]. Additional baseline clinical laboratory values of interest (total bilirubin, alkaline phosphatase, albumin, S-aspartate aminotransferase, and S-alanine aminotransferase) were explored graphically using the base population pharmacokinetic model (without covariates) to examine potential associations with pharmacokinetic parameters (CL and Vc; Supplemental Table 1). Further, the effect of coadministration of AMG 386 with paclitaxel compared with AMG 386 monotherapy was assessed after the selection of physiologic covariates.
Continuous covariates such as body weight were included in the structural model using the following equation:
where θ i is the value of a pharmacokinetic parameter for the ith individual; θTypical is the typical value of the pharmacokinetic parameter for an individual having the reference value of the covariate in the population (e.g., 70 kg for body weight); Cov i is the value of the covariate for the ith individual; Covreference value is the median of the covariate among patients, and θeff is the value of the covariate effect on the pharmacokinetic parameter.
The evaluation of a categorical covariate effect (e.g., sex) on typical values of pharmacokinetic parameters was described as follows:
The exposure measure in the exposure–response analysis was the steady-state area under the concentration-versus-time curve (AUCss), which was assessed based on individual CL values obtained with an empirical Bayesian post hoc estimate from the final population pharmacokinetic model (Supplemental Table 1) [21].
Exposure–response analysis
Kaplan–Meier curves for PFS were calculated for patients with AMG 386 AUCss ≥ 75th percentile (high exposure) and AUCss < 75th percentile (low exposure) and for placebo-treated patients. Univariate Cox regression models of AUCss by PFS were used to characterize the exposure-PFS curve. These models assumed a linear relationship between exposure and the logarithm of the relative risk. AUCss for patients who received placebo plus weekly paclitaxel was set to zero. Because the relationship between exposure and PFS may not have been linear over the entire range of study exposures, the following subsets were analyzed: placebo and AMG 386 3 mg/kg QW combined; AMG 386 3 mg/kg and 10 mg/kg QW combined; and AMG 386 10 mg/kg QW.
Multivariate Cox regression models were used to evaluate the effect of potential confounding factors on the exposure-PFS curve for the AMG 386 10 mg/kg dose. They included those affecting AMG 386 exposure (CrCL, age, and body weight), prognostic factors (Gynecological Oncology Group performance status, tumor type, histology, platinum sensitivity, progression on or within 6 months of previous chemotherapy regimen, and liver metastases), and baseline laboratory values (serum CA-125, albumin, alkaline phosphatase, S-aspartate aminotransferase, S-alanine aminotransferase, creatinine, lactate dehydrogenase, and potassium). A forward selection algorithm was used to identify a multivariate model with up to three variables using only data from the placebo group, a model that was associated with PFS in the absence of AMG 386 exposure; AUCss was added to this model and estimated for patients in the 10 mg/kg QW treatment arm to evaluate the effect of AMG 386 exposure on PFS when adjusting for factors with an AMG 386-absent association with PFS.
A descriptive analysis was conducted to evaluate trends in the incidence of severe (grade ≥3) AEs among patients with AMG 386 AUCss ≥ 75th percentile and < 75th percentile.
Simulation and dose assessment
To predict PFS at different doses, a parametric survival model that related estimated AUCss to PFS was developed using data from all three treatment arms in the phase 2 study. The survival function that best described the observed PFS distribution was selected from normal, log-normal, Weibull, logistic, log-logistic, and exponential functions using the Akaikie Information Criterion and diagnostic plots [9, 10, 24]. Estimation of model parameters was performed using the CensorReg function in S-PLUS (version 7.0; Insightful Corporation, Seattle, WA).
Evaluation of the selected model was conducted by simulating PFS values from 1,000 trials (replicates) and comparing the actual and simulated PFS curves (median and 95% CI) across all three treatment arms. To evaluate the AUCss distribution at different doses, random selections of 1,000 hypothetical patients were resampled (bootstrapped with replacements) from a total of 160 patients in the phase 2 study. Individual plasma concentration-versus-time profiles were simulated using the final population pharmacokinetic model including fixed- and random-effect parameters.
The population pharmacokinetic and survival models were used to simulate AUCss at AMG 386 doses of 0, 3, 10, and 15 mg/kg QW and to predict PFS across 1,000 replicates of a simulated 1,000-patient study, respectively. The objective of this analysis was to predict PFS following the treatment with various doses of AMG 386. Uncertainty in the AUCss-PFS model estimation was accounted for by sampling parameter estimates as part of the simulation process using a previously described method [9, 10]. In addition to the graphic comparison of the actual and predicted PFS curves, statistical estimates, including median PFS for each treatment arm and hazard ratio (HR) relative to placebo, were estimated and compared with the actual values.
Results
Patients
The final database for pharmacokinetic analysis consisted of 1,275 evaluable serum AMG 386 concentration assessments, of which 690 were from the first-in-human phase 1 study (from 32 patients with solid tumors), and 585 were from the phase 2 study (from 109 patients with recurrent ovarian cancer; Supplemental Table 2). Selected baseline characteristics of the two patient populations are summarized in Supplemental Table 3. Results from the primary analysis of the phase 2 study have been reported previously [17].
Progression-free survival (the primary endpoint) in the phase 2 study was 7.2 months (95% CI, 5.3–8.1) in the AMG 386 10 mg/kg QW dose group (HR, 0.76; 95% CI, 0.49–1.18; P = 0.225) and 5.7 months (95% CI, 4.6–8.0) in the 3 mg/kg group (HR, 0.75; 95% CI, 0.48–1.17; P = 0.207), compared with 4.6 months (95% CI, 1.9–6.7) for placebo [17]. Results from Tarone’s test and dose-exposure analyses suggested a dose–response effect for PFS across the three arms (P = 0.037).
Population pharmacokinetic analysis
AMG 386 exhibited dose-linear pharmacokinetic properties between 0.3 and 30 mg/kg QW. Among the covariates assessed, CrCL (calculated based on the Cockcroft-Gault formula) had the most pronounced effect on CL. Across the range of CrCL values collected in these 2 studies, the relationship between CL and CrCL was approximately linear (R 2, 0.33; CL, 43 mL/h + 0.356 × CrCL) (Fig. 1). For Vc, body weight and sex were significant covariates (Supplemental Table 1). No significant trends were seen when laboratory values for albumin, alkaline phosphatase, total bilirubin, S-aspartate aminotransferase, and S-alanine aminotransferase were plotted against the interindividual variability of CL or Vc from the base model. Mean AMG 386 CL based on the estimated individual CL was similar across the phase 1 monotherapy study (0.075 ± 0.020 L/h) and the phase 2 combination study (0.084 ± 0.026 L/h), suggesting that concomitant administration of paclitaxel did not affect the pharmacokinetics of AMG 386. The typical CL (0.072 L/h) for AMG 386 was faster than for monoclonal antibodies [25, 26].

Population pharmacokinetic analysis shows a linear correlation between AMG 386 clearance (CL) and creatinine clearance (CrCL)
Exposure–response analysis
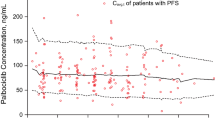
Univariate analysis demonstrated an exposure–response relationship between AUCss and PFS within each 1-unit increment in predicted AUCss (e.g., AUCss of 9 vs. 8 mg h/mL) for all patients who received AMG 386 (HR, 0.97; P = 0.097). Similar relationships were seen in subset analyses of the placebo and AMG 386 3 mg/kg arms combined (HR, 0.90; P = 0.121) and the AMG 386 3 mg/kg and 10 mg/kg arms combined (HR, 0.97; P = 0.23) as well as the 10 mg/kg arm alone (HR, 0.89; P = 0.069). The relationship between AUCss and PFS remained (adjusted HR, 0.87, P = 0.045) in the 10 mg/kg arm after adjusting for potential confounding factors (platinum sensitivity, baseline CA-125, and progression on or within 6 months of previous chemotherapy; Table 1), indicating that the trend was likely due to a true exposure–response relationship rather than a correlation between a potential confounder and AUCss. These results also suggested that the maximum effect on PFS was not achieved with a dose of 10 mg/kg QW.
An AMG 386 AUCss of 9.6 mg h/mL (the 75th percentile of median AUCss among patients administered AMG 386) was selected as a cut point for further analysis. Although the effect of higher AMG 386 AUCss on clinical benefit was also analyzed, 9.6 mg h/mL was selected to ensure that a sufficient number of patients were included in safety and efficacy evaluations. Figure 2 shows Kaplan–Meier curves for estimated PFS when patients were grouped by the dose of AMG 386 administered or by AUCss. Patients with high exposure to AMG 386 (AUCss ≥ 9.6 mg h/mL) showed a more marked separation in PFS curves versus placebo, compared with patients in the low-exposure group (AUCss < 9.6 mg h/mL). Median PFS was 4.6 months (95% CI, 1.9–6.7) among placebo patients (n = 55), 8.1 months (95% CI, 6.4–10.8) among patients with AUCss ≥ 9.6 mg h/mL (n = 26; HR vs. placebo, 0.67 [95% CI, 0.40–1.14]), and 5.7 months (95% CI, 4.6–7.5) among patients with AUCss < 9.6 mg h/mL (n = 79; HR vs. placebo, 0.81 [95% CI, 0.55–1.21]).

Kaplan–Meier plots of progression-free survival (PFS; per Response Evaluation Criteria in Solid Tumors, clinical progression or CA-125 progression, or death) stratified by treatment arm (a) or exposure to ≥9.6 or <9.6 mg h/mL (b). AUCss, steady-state area under the concentration-versus-time curve; QW, once weekly
Exposure-safety analysis
A descriptive exposure-safety analysis did not reveal any clinically relevant differences in the incidence of AEs among patients with high (AUCss ≥ 9.6 mg h/mL) and low (AUCss < 9.6 mg h/mL) AMG 386 exposure (Table 2). No relationship between AUCss and grade ≥ 3 adverse events was observed. Differences were seen between some AEs (e.g., peripheral neuropathy, arthralgia), but the study was not powered to detect statistically significant differences in the incidence of individual AEs. Additionally, these toxicities are commonly associated with paclitaxel administration and, therefore, may be attributed to longer duration of chemotherapy administration in this patient subset rather than to AMG 386 administration.
Simulation and dose assessment
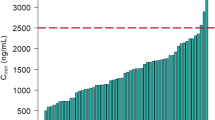
Figure 3 shows the distributions of the predicted AUCss with AMG 386 at a dose of 15 mg/kg QW and the actual AUCss data that were seen with administration of 3 or 10 mg/kg QW (phase 2 combination study), or 30 mg/kg QW (phase 1 monotherapy study). At a dose of 15 mg/kg QW, >90% of patients were predicted to achieve an AUCss ≥ 9.6 mg h/mL, which is well below the median AUCss (24.9 mg h/mL) reported for a dose of 30 mg/kg.

Distribution of observed steady-state area under the concentration-versus-time curve (AUCss) following once-weekly (QW) doses of AMG 386 3 or 10 mg/kg (a), predicted AUCss following simulated AMG 386 15 mg/kg QW (b), and observed AUCss following AMG 386 30 mg/kg QW in the first-in-human study (c)
Among the various survival functions assessed, the Weibull distribution had the lowest Akaikie Information Criterion and, therefore, was used to describe the PFS data. The parametric model with Weibull distribution was subsequently used to characterize the simulated PFS data for AMG 386 doses of 0 (placebo), 3, 10, and 15 mg/kg in combination with paclitaxel. The Kaplan–Meier PFS curves predicted by the Weibull model for placebo and for 3 and 10 mg/kg QW were consistent with the actual PFS curves for patients in the corresponding treatment arms, as were the HRs for comparison with placebo (Fig. 4a–c). This finding confirms the adequacy of the simulated data and fitted parametric survival model, and the consistency of the simulated data with outcomes among patients with AUCss ≥ 9.6 mg h/mL. Additionally, the predicted Kaplan–Meier PFS curve for an AMG 386 dose of 15 mg/kg QW was similar to the actual curve based on data from patients in the AUCss ≥ 9.6 mg h/mL group (Fig. 4d). The predicted median PFS at an AMG 386 dose of 15 mg/kg QW was 8.2 months (95% CI, 5.4–11.9 months; HR, 0.56 [95% CI, 0.31–1.07]).

Predicted versus observed Kaplan–Meier estimates of progression-free survival (PFS). a Paclitaxel plus placebo; b AMG 386 3 mg/kg once weekly (QW); c AMG 386 10 mg/kg QW; d AMG 386 15 mg/kg QW (data source: AUCss ≥ 9.6 mg h/mL; n = 26). Graphs depict 1,000 replicates of 1,000 patients for each dose
Discussion
This study describes an important but infrequently used application of population pharmacokinetic/pharmacodynamic modeling to guide dose selection for phase 3 studies of an antiangiogenic agent. Attrition rates for investigational cancer therapeutics are high [27]. Regulatory guidance and the published literature suggest that the integration of pharmacokinetic, pharmacodynamic, and clinical endpoint data may better inform future study design and help maximize the risk–benefit profile for therapeutics [28–30]. In particular, exposure–response modeling may aid in the rational selection of doses for further investigation [31, 32]. The failure of some cancer therapeutics in development may be due to the conventional approach to dose selection, which primarily focuses on the determination of the maximum tolerated dose [33], whereas identification of an “optimal biologic dose” may be more appropriate for targeted agents [34]. Consistent with this approach, a number of recent phase 1 studies have used exposure–response modeling to assess the relationship between exposure and a marker of biologic activity to facilitate dose selection [35–39]. However, because these markers have not been clinically validated, the appropriateness of such analyses for use in dose selection has been uncertain [40].
The present study was a prospectively planned pharmacokinetic/pharmacodynamic analysis that assessed the relationship between exposure (AUCss) and a key clinical outcome (PFS) to guide dose selection for phase 3 studies of AMG 386 in recurrent ovarian cancer. The population pharmacokinetic part of the analysis revealed that CrCL, a measure of renal function, appears to be a significant covariate for AMG 386 CL. The relationship suggests that renal disposition may play a role in the elimination of AMG 386, which, at a size of approximately 65 kDa, is a fairly large molecule. Renal clearance is uncommon for biologic therapeutics, such as monoclonal antibodies, and, to our knowledge, has not been described previously. Estimated glomerular filtration rate (calculated using the Modification of Diet in Renal Disease, MDRD, method), which is another measure of renal function, also showed a significant effect on AMG 386 CL (data not shown). This further supports our finding that the kidney may be implicated in the elimination of AMG 386. However, the exact mechanism of the effect of CrCL on the CL of AMG 386 remains uncertain and warrants further investigation.
Exposure–response analysis revealed a robust relationship between AMG 386 exposure and PFS, suggesting that maximum clinical benefit was not reached at a dose of 10 mg/kg QW. The exposure-PFS relationship remained after adjusting for potential confounding factors in the multivariate analysis. However, given the relatively small sample size of the phase 2 study, other unknown confounding factors may have introduced an unidentified bias. Using the results from the simulations based on the parametric survival model, an AMG 386 dose of 15 mg/kg QW in combination with cytotoxic chemotherapy has been proposed for phase 3 studies in patients with recurrent ovarian cancer (TRINOVA-1 [ClinicalTrials.gov, NCT01204749] and TRINOVA-2 [ClinicalTrials.gov, NCT01281254]). Although the toxicity of this dose when combined with paclitaxel has not yet been directly tested, the exposure-safety analysis presented here suggests that 15 mg/kg of AMG 386 will have a similar safety profile as the 10 mg/kg dose. There were no marked differences in the incidence of grade ≥ 3 AEs between patients with high and low AMG 386 exposure, and the primary analysis did not show any apparent dose-related trends in toxicity when comparing 3 and 10 mg/kg QW administered in combination with paclitaxel [17]. In the phase 1 study, 30 mg/kg QW (the maximum tested dose) was well tolerated as monotherapy [18].
Exposure–response relationships appear to be influenced by a number of factors, which can complicate efforts to identify an optimal biological exposure (OBE) and optimal biological dose (OBD) for a given anticancer agent. OBDs and OBEs from monotherapy dose-escalation studies in mixed solid tumors may not translate into later-stage studies (monotherapy or combination therapy) of single tumor types. For example, not all clinical studies of the anti-VEGF-A antibody bevacizumab have shown a consistent dose–response relationship, suggesting that different optimum doses may be needed for different tumor types or disease characteristics [41]. Furthermore, an agent’s OBE may differ between in vitro models and clinical studies. The AMG 386 OBE for antitumor efficacy in xenograft models [16] appeared to be lower than that identified in the phase 2 ovarian cancer study. This may reflect differences in receptor occupancy across species, which has been observed in other contexts [42].
Our study demonstrates the use of a novel model-based approach to dose selection for a phase 3 study of an investigational targeted therapy. Applying this technique to the decision-making process in the development of anticancer agents, for which dose-ranging studies are rarely performed [8], provides important opportunities. Integrating results from preclinical pharmacokinetic, pharmacologic, and toxicity studies into appropriate models can guide the design of early clinical studies and inform the interpretation of its results, thus supporting the fast transition of a promising molecule from discovery into the clinic. Go/no-go decisions during continued clinical development and dose selection for late-stage studies can also be successfully supported by modeling applications. Thus, quantitative (such as pharmacokinetic/pharmacodynamic and/or exposure–response) modeling and simulations can guide each step of a clinical development plan from early discovery through pivotal phase 3 studies [1]. However, this approach is often limited because it requires early integration of pharmacometric scientists in the clinical decision-making process as well as the timely development of relevant models.
In summary, our study demonstrates how exposure–response analyses of phase 2 study data and the application of pharmacokinetic/pharmacodynamic models can assist in the selection of doses for subsequent phase 3 studies of an antiangiogenic therapeutic.
References
Wetherington JD, Pfister M, Banfield C, Stone JA, Krishna R, Allerheiligen S, Grasela DM (2010) Model-based drug development: strengths, weaknesses, opportunities, and threats for broad application of pharmacometrics in drug development. J Clin Pharmacol 50:31S–46S
Gobburu JV (2010) Pharmacometrics 2020. J Clin Pharmacol 50:151S–157S
Goffin J, Baral S, Tu D, Nomikos D, Seymour L (2005) Objective responses in patients with malignant melanoma or renal cell cancer in early clinical studies do not predict regulatory approval. Clin Cancer Res 11:5928–5934
Ratain MJ (2005) Phase II oncology trials: let’s be positive. Clin Cancer Res 11:5661–5662
Appels NM, Bolijn MJ, Chan K, Stephens TC, Hoctin-Boes G, Middleton M, Beijnen JH, de Bono JS, Harris AL, Schellens JH (2008) Phase I pharmacokinetic and pharmacodynamic study of the prenyl transferase inhibitor AZD3409 in patients with advanced cancer. Br J Cancer 98:1951–1958
Karrison TG, Maitland ML, Stadler WM, Ratain MJ (2007) Design of phase II cancer trials using a continuous endpoint of change in tumor size: application to a study of sorafenib and erlotinib in non small-cell lung cancer. J Natl Cancer Inst 99:1455–1461
Workman P (2002) Challenges of PK/PD measurements in modern drug development. Eur J Cancer 38:2189–2193
Ratain MJ, Humphrey RW, Gordon GB, Fyfe G, Adamson PC, Fleming TR, Stadler WM, Berry DA, Peck CC (2008) Recommended changes to oncology clinical trial design: revolution or evolution? Eur J Cancer 44:8–11
Claret L, Girard P, Hoff PM, Van Cutsem E, Zuideveld KP, Jorga K, Fagerberg J, Bruno R (2009) Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol 27:4103–4108
Wang Y, Sung C, Dartois C, Ramchandani R, Booth BP, Rock E, Gobburu J (2009) Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther 86:167–174
Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ (2010) Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol 66:357–371
Jimeno A, Rudek MA, Kulesza P, Ma WW, Wheelhouse J, Howard A, Khan Y, Zhao M, Jacene H, Messersmith WA, Laheru D, Donehower RC, Garrett-Mayer E, Baker SD, Hidalgo M (2008) Pharmacodynamic-guided modified continuous reassessment method-based, dose-finding study of rapamycin in adult patients with solid tumors. J Clin Oncol 26:4172–4179
Lu J-F, Claret L, Sutjandra L, Kuchimanchi M, Melara R, Bruno R, Sun Y-N (2010) Population pharmacokinetic/pharmacodynamic modeling for the time course of tumor shrinkage by motesanib in thyroid cancer patients. Cancer Chemother Pharmacol 66:1151–1158
Guidance for industry end-of-phase 2A meetings (2009). United States Food and Drug Administration. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm079690.pdf. Accessed 24 Aug 2009
US Food and Drug Administration (2006) Critical path opportunities report. US Department of Health and Human Services, Washington
Oliner J, Min H, Leal J, Yu D, Rao S, You E, Tang X, Kim H, Meyer S, Han SJ, Hawkins N, Rosenfeld R, Davy E, Graham K, Jacobsen F, Stevenson S, Ho J, Chen Q, Hartmann T, Michaels M, Kelley M, Li L, Sitney K, Martin F, Sun JR, Zhang N, Lu J, Estrada J, Kumar R, Coxon A, Kaufman S, Pretorius J, Scully S, Cattley R, Payton M, Coats S, Nguyen L, Desilva B, Ndifor A, Hayward I, Radinsky R, Boone T, Kendall R (2004) Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell 6:507–516
Karlan BY, Oza AM, Richardson GE, Provencher DM, Hansen VL, Buck M, Chambers SK, Ghatage P, Pippitt Jr. CH, Brown III JV, Covens A, Nagarkar RV, Davy M, Leath III CA, Nguyen H, Stepan DE, Weinreich DM, Tassoudji M, Sun YN, Vergote IB (2011) A randomized, double-blind, placebo-controlled phase 2 study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian cancer. J Clin Oncol. doi:10.1200/JCO.2010.34.3178
Herbst RS, Hong D, Chap L, Kurzrock R, Jackson E, Silverman JM, Rasmussen E, Sun YN, Zhong D, Hwang YC, Evelhoch JL, Oliner JD, Le N, Rosen LS (2009) Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J Clin Oncol 27:3557–3565
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216
Rustin GJ, Marples M, Nelstrop AE, Mahmoudi M, Meyer T (2001) Use of CA-125 to define progression of ovarian cancer in patients with persistently elevated levels. J Clin Oncol 19:4054–4057
Beal SL, Sheiner LB, Boeckmann AJ (eds) (1989–1998) NONMEM users guides. Icon Development Solutions, Ellicott City
Mandema JW, Verotta D, Sheiner LB (1992) Building population pharmacokinetic–pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm 20:511–528
Cockcroft DW, Gault MH (1976) Prediction of creatinine clearance from serum creatinine. Nephron 16:31–41
Marubini E, Grazia Valsecchi M (1995) Analysing survival data from clinical trials and observational studies. Wiley, Chichester
Lu JF, Bruno R, Eppler S, Novotny W, Lum B, Gaudreault J (2008) Clinical pharmacokinetics of bevacizumab in patients with solid tumors. Cancer Chemother Pharmacol 62:779–786
Bruno R, Washington CB, Lu JF, Lieberman G, Banken L, Klein P (2005) Population pharmacokinetics of trastuzumab in patients with HER2+ metastatic breast cancer. Cancer Chemother Pharmacol 56:361–369
Rowinsky EK (2004) Curtailing the high rate of late-stage attrition of investigational therapeutics against unprecedented targets in patients with lung and other malignancies. Clin Cancer Res 10:4220s–4226s
Gombar C, Loh E (2007) Learn & confirm. Drug Discov Dev 10:22–27
European Medicines Agency (2005) The European Medicines Agency road map to 2010: preparing the ground for the future. London
US Food and Drug Administration (2004) Innovation or stagnation: challenge and opportunity on the critical path to new medical products. US Department of Health and Human Services. US Food and Drug Administration, Rockville
Agoram BM (2009) Use of pharmacokinetic/pharmacodynamic modelling for starting dose selection in first-in-human trials of high-risk biologics. Br J Clin Pharmacol 67:153–160
Rajman I (2008) PK/PD modelling and simulations: utility in drug development. Drug Discov Today 13:341–346
Jain RK, Lee JJ, Hong D, Markman M, Gong J, Naing A, Wheler J, Kurzrock R (2010) Phase I oncology studies: evidence that in the era of targeted therapies patients on lower doses do not fare worse. Clin Cancer Res 16:1289–1297
Adjei AA (2006) What is the right dose? The elusive optimal biologic dose in phase I clinical trials. J Clin Oncol 24:4054–4055
Puchalski T, Prabhakar U, Jiao Q, Berns B, Davis HM (2010) Pharmacokinetic and pharmacodynamic modeling of an anti-interleukin-6 chimeric monoclonal antibody (siltuximab) in patients with metastatic renal cell carcinoma. Clin Cancer Res 16:1652–1661
Tanaka C, O’Reilly T, Kovarik JM, Shand N, Hazell K, Judson I, Raymond E, Zumstein-Mecker S, Stephan C, Boulay A, Hattenberger M, Thomas G, Lane HA (2008) Identifying optimal biologic doses of everolimus (RAD001) in patients with cancer based on the modeling of preclinical and clinical pharmacokinetic and pharmacodynamic data. J Clin Oncol 26:1596–1602
O’Donnell A, Faivre S, Burris HA III, Rea D, Papadimitrakopoulou V, Shand N, Lane HA, Hazell K, Zoellner U, Kovarik JM, Brock C, Jones S, Raymond E, Judson I (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol 26:1588–1595
Li J, Jameson MB, Baguley BC, Pili R, Baker SD (2008) Population pharmacokinetic-pharmacodynamic model of the vascular-disrupting agent 5, 6-dimethylxanthenone-4-acetic acid in cancer patients. Clin Cancer Res 14:2102–2110
Zandvliet AS, Karlsson MO, Schellens JH, Copalu W, Beijnen JH, Huitema AD (2010) Two-stage model-based clinical trial design to optimize phase I development of novel anticancer agents. Invest New Drugs 28:61–75
Sleijfer S, Wiemer E (2008) Dose selection in phase I studies: why we should always go for the top. J Clin Oncol 26:1576–1578
Bergsland E, Dickler MN (2004) Maximizing the potential of bevacizumab in cancer treatment. Oncologist 9(Suppl 1):36–42
Lowe PJ (2010) Applying physiological and biochemical concepts to optimize biological drug development. Clin Pharmacol Ther 87:492–496
Acknowledgments
The authors thank Ali Hassan, PhD (Complete Healthcare Communications, Inc., Chadds Ford, PA), and Emil Samara, PhD (PharmaPolaris, Danville, CA), whose work was funded by Amgen Inc. (Thousand Oaks, CA), and Beate D. Quednau, PhD (Amgen Inc.), for assistance in the preparation of this manuscript. This study was supported by Amgen Inc.
Conflict of interest
Jian-Feng Lu, Erik Rasmussen, Lynn Navale, Mita Kuchimanchi, Rebeca Melara, Daniel E. Stepan, David M. Weinreich, and Yu-Nien Sun are employees of and shareholders in Amgen Inc. Beth Y. Karlan has received research funding from Amgen Inc. Ignace B. Vergote has no conflicts to declare.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Clinical trial registration: ClinicalTrials.gov registration numbers: NCT00479817 and NCT00102830.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lu, JF., Rasmussen, E., Karlan, B.Y. et al. Exposure–response relationship of AMG 386 in combination with weekly paclitaxel in recurrent ovarian cancer and its implication for dose selection. Cancer Chemother Pharmacol 69, 1135–1144 (2012). https://doi.org/10.1007/s00280-011-1787-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00280-011-1787-5