
Abstract
Immune thrombocytopenia (ITP) is an autoimmune disease primarily characterized by increased clearance of auto-antibody-sensitized platelets by Fc-receptor-bearing macrophages in the spleen and liver. It has been classically accepted that antibody-mediated platelet destruction is Fc dependent. Recent studies, however, may also indicate the involvement of Fc-independent pathways of platelet destruction. Current treatment options work by immunosuppression (e.g., corticosteroids), immunomodulation (e.g., IVIg and anti-D), or removal of the platelet destruction site (splenectomy) in ITP. This review will discuss the mechanisms of action of these and other treatments for ITP.
Similar content being viewed by others

Introduction
Immune thrombocytopenia (ITP) is an autoimmune disease generally characterized by increased clearance of auto-antibody-sensitized platelets by Fc-receptor (FcR)-bearing macrophages [1, 2]. In addition, decreased platelet production is partly responsible for the reduced platelet count in ITP [3]. The disorder varies in its severity and clinical presentation. The terminology used to describe ITP and its treatment outcomes has been recently revised and standardized and an international consensus on the investigation and management of ITP has also recently been published [4, 5]. Recently diagnosed childhood ITP is often preceded by viral infection. Most childhood cases resolve spontaneously, regardless of whether treatment is given. Up to 20% become chronic with the ITP persisting for more than 12 months. In contrast to childhood ITP, ITP in adults typically occurs with an unknown cause and is not generally associated with an underlying disorder [1].
It is currently accepted that antibody-mediated platelet destruction is Fc dependent. Recent studies however, may also indicate the involvement of Fc-independent pathways of platelet destruction [6–10]. Current treatment options work by immunosuppression (e.g., corticosteroids), immunomodulation (e.g., IVIg and anti-D), or removal of the platelet destruction site (splenectomy). The heterogeneity of ITP etiology makes the disorder unpredictable in its requirement for, and response to, therapy.
Mechanisms of platelet destruction in ITP
In order to understand platelet recovery with treatment, the causes of ITP need to be defined. A review of the literature indicates the possibility of more than one mechanism of platelet destruction. In addition to the well-documented Fc-dependent mechanism of platelet elimination, recent studies have highlighted the contribution of Fc-independent pathways.
Fc-dependent mechanisms
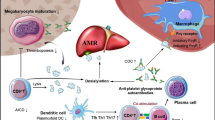
The basic underlying pathophysiology of ITP is the targeting of platelets, by Ag-specific IgG antibodies, followed by Fcγ-receptor (FcγR)-mediated phagocytosis in the spleen and liver [11]. The low-affinity FcγRIIA and FcγRIII have been speculated to be keys in FcγR-mediated platelet elimination in ITP [1, 12]. IgG-bound platelets engage FcγRs via its Fc portion, initiating cytosolic signaling pathways that culminate in their uptake and destruction [13].
The IgG repertoire present affects the course of ITP. A study characterizing anti-glycoprotein (GP) IIb/IIIa antibodies in ITP patients found that the IgG subclass distribution of these antibodies differed among patients [14]. Some patients had anti-GPIIb/IIIa of multiple subclasses, while evidence of IgG subclass restrictions was found in others. Patients varied in the severity of their ITP, with those producing IgG4 having less severe thrombocytopenia [14].
The identities of the auto-antibody targets have also been shown to vary responses to therapy. Most anti-platelet antibodies are directed against GPIIb/IIIa and/or GPIb/IX. Previous studies have shown that the immune thrombocytopenia caused by anti-GPIb/IX antibodies may be less responsive to IVIg therapy (discussed later) [9, 15].
Fc-independent mechanisms
Several lines of work support Fc-independent platelet elimination in ITP. Nieswandt and colleagues determined that both intact monoclonal anti-GPIb/IX antibody and their F(ab)2 fragments can induce a decrease in the platelet count in mice [16]. Interestingly, anti-GPIIb/IIIa antibodies can also cause thrombocytopenia by an Fc-independent mechanism in some settings. Zhang et al. showed that the hepatitis C virus (HCV) core protein 1 may induce thrombocytopenia by molecular mimicry [8]. Antibodies specific for the amino acid sequence 49-66 of GPIIIa, cross-reacted with four nonconserved peptides from the HCV core protein 1. These anti-GPIIIa49-66 antibodies induced complement-independent platelet fragmentation by activating the platelet 12-lipoxygenase, producing 12(S)-hydroxyeicosatetraenoic acid, which then activates the platelet NADPH oxidase pathway, generating reactive oxygen species (ROS) [17]. ROS release causes the platelet to fragment. Such a mechanism of platelet destruction has been reported in thrombocytopenic HIV patients. Wright et al. previously showed that childhood ITP preceded by varicella zoster virus (VZV) infection may be due to molecular mimicry by viral proteins [10]. It was reported that anti-VZV antibodies could bind platelets and activate complement factors [10]
Some studies have provided support for complement involvement in ITP. The membrane attack complex C5-9 can be induced by anti-platelet antibodies in ITP [18]. Additionally, complement proteins C3 and C4 associated with platelet-complexed IgG have been found to shorten platelet lifespan. Furthermore, platelets themselves express complement receptors and regulatory proteins and have been shown to bind immune complexes [18].
T cell contributions to the pathogenesis of ITP have also been described. Olsson and colleagues showed that some chronic ITP patients possessed CD3+CD8+ T cells that mediated lysis of autologous platelets [6]. They also observed increased expression of Th1-associated cytokines including interferon-γ, and cytotoxic molecules such as Apo-l/Fas, granzymes A and B, and perforin [6]. Further confirmation of cytotoxic T cell-mediated involvement in ITP was provided by Zhang et al. [7]. They reported effector CD8+ T cells that mediated a low level of platelet lysis in some ITP patients. FasL and TNF-α expression and mRNA levels of granzyme B and perforin in these CD8+ T cells were increased [7]. Altogether, the implication of such varied platelet destruction mechanisms reflects the complex heterogeneity of ITP.
Mechanisms of platelet recovery after therapy
No universally effective therapeutic regiment for ITP exists. Traditional therapies often do not provide sustained benefits and unforeseen adverse effects are a concern. In recent years, ITP treatment has advanced with the development of newer, promising therapies.
Corticosteroids
Corticosteroids are pharmacological derivatives of the naturally occurring glucocorticoid (GC) family of steroid hormones. GCs bind cytoplasmic GC receptors found in most cell types [19]. The GC-receptor complex translocates to the nucleus where it regulates transcription of a network of genes. Corticosteroids decrease production and inhibit the action of a multitude of cytokines. They also promote T cell apoptosis, suppress their proliferation, and can also inhibit T-cell receptor signaling via a non-GC receptor effect [19]. Because of widespread GC receptor expression and the large number of genes regulated by corticosteroids, its use comes with complex adverse effects involving multiple organs and includes increased chances of infection, growth impairment in children, weight gain, bone mineral loss, and fluid retention [19].
IVIg
IVIg is a preparation of human polyclonal IgG made by pooling plasma from thousands of healthy blood donors [2]. IVIg is typically recommended for ITP cases unresponsive to corticosteroid therapy. Doses of 1-2 g/kg body weight are most often used to treat ITP. The half-life of administered IVIg is roughly 3 weeks. IVIg is a costly therapy and as IVIg is a blood product, there is always the potential for infectious disease transmission as well as transmission of non-conventional infectious agents.
IVIg has been shown to exert a range of immunomodulatory effects, the mechanisms of which are not fully understood. It has been theorized that IVIg functions by saturating FcγRs on macrophages, changing FcγR expression, increasing the catabolic rate of pathogenic antibodies via saturation of the neonatal FcR, altering the cytokine profile, providing anti-idiotypic antibodies, and modulating dendritic cell (DC) maturation, as well as affecting T and B cell function [12, 20–25]. A role for increased expression of the inhibitory FcγRIIB on macrophages has also been implicated in IVIg action [26]. Recent work from our laboratory and others support a role for dendritic cells in IVIg action. We showed that IVIg-treated splenic CD11c+ DCs could prevent the thrombocytopenia seen in a murine model of ITP [24]. Bayry et al. observed that IVIg inhibits in vitro DC activation, down-regulating surface co-stimulatory molecules and abrogating auto-reactive T cell activation [23]. Furthermore, a recent study by Anthony and colleagues implicate a role for SIGN-R1 (specific ICAM-3 grabbing non-integrin-related 1) in murine ITP. They found that its expression was required for protective IVIg effects and suggested that binding of IVIg may occur via the 2,6-sialylated Fc region of IgG [25]. It is interesting to note that SIGN-R1 is an orthologue of human DC-SIGN, which is found on dendritic cells in humans and is speculated to play a role in self-antigen recognition and tolerization. In summary, the mechanism(s) of IVIg action in ITP remains to be confirmed.
Anti-D
Polyclonal anti-D consists of IgG against the RhD antigen on RBCs. It is prepared from the plasma of RhD negative individuals immunized against the D antigen. Anti-D is considered to be only effective in RhD positive, nonsplenectomized individuals. In rare cases, patients given anti-D may develop potentially life-threatening intravascular hemolysis. The standard dose of anti-D is 50-75 µg/kg body weight given by intravenous infusion [27, 28].
Anti-D is presumed to work by competitive engagement of FcγRs by anti-D-coated RBCs, thereby blocking phagocytosis of antibody-sensitized platelets in the spleen [29, 30]. Interestingly however, a previous study showed that anti-D administration in ITP patients was associated with a decrease in anti-GPIIb/IIIa auto-antibodies [31]. Furthermore, work from our laboratory discovered that treatment of a murine ITP model with monoclonal anti-RBC antibodies reversed thrombocytopenia and downregulated the expression of the activating FcγRIIIA on splenic macrophages, independent of the inhibitory FcγRIIB [32]. These studies and others may suggest the possibility of additional mechanisms by which anti-D prevents platelet destruction.
Helicobacter pylori eradication
Recent studies have reported a potential contribution of H. pylori infection in some older ITP patients, although reports of H. pylori prevalence in ITP cases are different between countries [33, 34]. A study by Asahi et al. observed that H. pylori eradication lowered phagocytic capacity and elevated inhibitory FcγRIIB expression of monocytes in H. pylori-positive ITP patients [35]. H. pylori eradication shows promise as a cost-effective ITP therapy for some patients, particularly in countries prevalent in H. pylori infection.
Other treatments
The spleen is the primary site of platelet destruction and anti-platelet antibody production [11]. Splenectomy may be performed in patients unresponsive to first-line medications. Roughly two-thirds of patients show complete remission; however, a small percentage will relapse and splenectomy carries risks for infection, especially in young children.
Rituximab is a monoclonal antibody against CD20, which is present on all mature B lymphocytes. Its use results in sustained B cell depletion, decreased anti-platelet antibodies, rectification of ITP-associated aberrations in the T cell compartment including increased Th1/Th2 and Tc1/Tc2 ratios, inappropriate T cell apoptosis, and altered regulatory T cells [36, 37]. Long-term safety data are not available for rituximab and side effects have been observed including reactivation of hepatitis B in hepatitis B carriers.
Recent studies have introduced several thrombopoeitin (TPO) mimetics including romiplostim and eltrombopag as promising therapeutic agents for ITP. The peptibody romisplostim and the nonpeptide eltrombopag work by engaging the TPO receptor and inducing megakaryocyte and platelet production via the JAK2 and STAT5 kinase pathways [38, 39]. Adverse effects, mild and severe, have been reported [40, 41]. Long-term effects of both drugs are currently unknown [42].
Other drugs used to treat ITP include azathioprine, cyclosporin A, vinca alkaloids, and cyclophosphamide [27]. Their use is associated with severe adverse side effects such as neuropathy, nephrotoxicity, and leukemias [27]. These are not used as first-line therapy.
A pilot study by Podolanczuk et al. showed that the orally available Syk kinase inhibitor R406 was effective in increasing and maintaining platelet counts in roughly 50% of chronic refractory ITP patients. It is presumed to block the signaling pathway downstream of FcγR ligation by antibody-sensitized platelets [43].
Conclusions
ITP is a complex disease that is not fully understood. As such, there is no set therapeutic regimen to treat the disorder. Rigorous research has led to the identification of both Fc-dependent and independent pathways of platelet destruction in ITP. The improved understanding of ITP etiology has allowed the development of novel, more effective therapies to add to the current arsenal of mechanistically diverse treatments. Taken together, there is an imminent need for clearly defining platelet elimination mechanisms in ITP as well as platelet recovery with therapy.
References
Crow AR, Lazarus AH (2003) Role of Fcgamma receptors in the pathogenesis and treatment of idiopathic thrombocytopenic purpura. J Pediatr Hematol Oncol 25(Suppl 1):S14–S18
Jin F, Balthasar JP (2005) Mechanisms of intravenous immunoglobulin action in immune thrombocytopenic purpura. Hum Immunol 66(4):403–410
Kuter DJ, Gernsheimer TB (2009) Thrombopoietin and platelet production in chronic immune thrombocytopenia. Hematol Oncol Clin North Am 23(6):1193–1211
Rodeghiero F et al (2009) Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 113(11):2386–2393
Provan D et al (2010) International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 115(2):168–186. doi:10.1182/blood-06-225565
Olsson B et al (2003) T-cell-mediated cytotoxicity toward platelets in chronic idiopathic thrombocytopenic purpura. Nat Med 9(9):1123–1124
Zhang F et al (2006) Cell-mediated lysis of autologous platelets in chronic idiopathic thrombocytopenic purpura. Eur J Haematol 76(5):427–431
Zhang W et al (2009) Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 113(17):4086–4093
Webster ML et al (2006) Relative efficacy of intravenous immunoglobulin G in ameliorating thrombocytopenia induced by antiplatelet GPIIbIIIa versus GPIbalpha antibodies. Blood 108(3):943–946
Wright JF et al (1996) Characterization of platelet-reactive antibodies in children with varicella-associated acute immune thrombocytopenic purpura (ITP). Br J Haematol 95(1):145–152
Kuwana M et al (2002) Spleen is a primary site for activation of platelet-reactive T and B cells in patients with immune thrombocytopenic purpura. J Immunol 168(7):3675–3682
Crow AR, Lazarus AH (2008) The mechanisms of action of intravenous immunoglobulin and polyclonal anti-d immunoglobulin in the amelioration of immune thrombocytopenic purpura: what do we really know? Transfus Med Rev 22(2):103–116
Aslam R et al (2007) Platelet and red blood cell phagocytosis kinetics are differentially controlled by phosphatase activity within mononuclear cells. Transfusion 47(11):2161–2168
Chan H et al (2003) The IgG subclasses of platelet-associated autoantibodies directed against platelet glycoproteins IIb/IIIa in patients with idiopathic thrombocytopenic purpura. Br J Haematol 122(5):818–824
Go RS, Johnston KL, Bruden KC (2007) The association between platelet autoantibody specificity and response to intravenous immunoglobulin G in the treatment of patients with immune thrombocytopenia. Haematologica 92(2):283–284
Nieswandt B et al (2000) Identification of critical antigen-specific mechanisms in the development of immune thrombocytopenic purpura in mice. Blood 96(7):2520–2527
Nardi M et al (2004) Complement-independent Ab-induced peroxide lysis of platelets requires 12-lipoxygenase and a platelet NADPH oxidase pathway. J Clin Invest 113(7):973–980
Hed J (1998) Role of complement in immune or idiopathic thrombocytopenic purpura. Acta Paediatr Suppl 424:37–40
Cole TJ (2006) Glucocorticoid action and the development of selective glucocorticoid receptor ligands. Biotechnology Ann Rev 12:269–300
Fehr J, Hofmann V, Kappeler U (1982) Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous gamma globulin. N Engl J Med 306(21):1254–1258
Berchtold P et al (1989) Inhibition of autoantibody binding to platelet glycoprotein IIb/IIIa by anti-idiotypic antibodies in intravenous gammaglobulin. Blood 74(7):2414–2417
Hansen RJ, Balthasar JP (2002) Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. Thromb Haemost 88(6):898–899
Bayry J et al (2003) Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood 101(2):758–765
Siragam V et al (2006) Intravenous immunoglobulin ameliorates ITP via activating Fc gamma receptors on dendritic cells. Nat Med 12(6):688–692
Anthony RM et al (2008) Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A 105(50):19571–19578
Samuelsson A et al (2001) Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 291(5503):484–486
Stasi R, Provan D (2004) Management of immune thrombocytopenic purpura in adults. Mayo Clin Proc 79(4):504–522
Newman GC et al (2001) A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 112(4):1076–1078
Salama A et al (1984) Treatment of autoimmune thrombocytopenic purpura with rhesus antibodies (anti-Rh0(D)]. Blut 49(1):29–35
Ambriz-Fernandez R et al (2002) Fc receptor blockade in patients with refractory chronic immune thrombocytopenic purpura with anti-D IgG. Arch Med Res 33(6):536–540
Boughton BJ et al (1994) Autoimmune thrombocytopenia: anti-glycoprotein IIb/IIIa auto antibodies are reduced after human anti-D immunoglobulin treatment. Autoimmunity 18(2):141–144
Song S et al (2005) Monoclonal antibodies that mimic the action of anti-D in the amelioration of murine ITP act by a mechanism distinct from that of IVIg. Blood 105(4):1546–1548
Stasi R et al (2009) Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 113(6):1231–1240
Franchini M et al (2007) Effect of Helicobacter pylori eradication on platelet count in idiopathic thrombocytopenic purpura: a systematic review and meta-analysis. J Antimicrob Chemother 60(2):237–246
Asahi A et al (2008) Helicobacter pylori eradication shifts monocyte Fcgamma receptor balance toward inhibitory FcgammaRIIB in immune thrombocytopenic purpura patients. J Clin Invest 118(8):2939–2949
Stasi R et al (2007) Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 110(8):2924–2930
Stasi R et al (2008) Analysis of regulatory T-cell changes in patients with idiopathic thrombocytopenic purpura receiving B cell-depleting therapy with rituximab. Blood 112(4):1147–1150
Erickson-Miller CL et al (2005) Discovery and characterization of a selective, nonpeptidyl thrombopoietin receptor agonist. Exp Hematol 33(1):85–93
Cohn CS, Bussel JB (2009) Romiplostim: a second-generation thrombopoietin agonist. Drugs Today (Barc) 45(3):175–188
Bussel JB et al (2007) Eltrombopag for the treatment of chronic idiopathic thrombocytopenic purpura. N Engl J Med 357(22):2237–2247
Kuter DJ et al (2008) Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 371(9610):395–403
Gernsheimer TB (2008) The pathophysiology of itp revisited: ineffective thrombopoiesis and the emerging role of thrombopoietin receptor agonists in the management of chronic immune thrombocytopenic purpura. Hematology Am Soc Hematol Educ Program 2008:219–226
Podolanczuk A et al (2009) Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood 113(14):3154–3160
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pang, S.J.Y., Lazarus, A.H. Mechanisms of platelet recovery in ITP associated with therapy. Ann Hematol 89 (Suppl 1), 31–35 (2010). https://doi.org/10.1007/s00277-010-0916-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00277-010-0916-2