
Abstract
Optimization of yields and productivities in reductive whole-cell biotransformations is an important issue for the industrial application of such processes. In a recent study with Escherichia coli, we analyzed the reduction of the prochiral β-ketoester methyl acetoacetate by an R-specific alcohol dehydrogenase (ADH) to the chiral hydroxy ester (R)-methyl 3-hydroxybutyrate (MHB) using glucose as substrate for the generation of NADPH. Deletion of the phosphofructokinase gene pfkA almost doubled the yield to 4.8 mol MHB per mole of glucose, and it was assumed that this effect was due to a partial cyclization of the pentose phosphate pathway (PPP). Here, this partial cyclization was confirmed by 13C metabolic flux analysis, which revealed a negative net flux from glucose 6-phosphate to fructose 6-phosphate catalyzed by phosphoglucose isomerase. For further process optimization, the genes encoding the glucose facilitator (glf) and glucokinase (glk) of Zymomonas mobilis were overexpressed in recombinant E. coli strains carrying ADH and deletions of either pgi (phosphoglucose isomerase), or pfkA, or pfkA plus pfkB. In all cases, the glucose uptake rate was increased (30–47%), and for strains Δpgi and ΔpfkA also, the specific MHB production rate was increased by 15% and 20%, respectively. The yield of the latter two strains slightly dropped by 11% and 6%, but was still 73% and 132% higher compared to the reference strain with intact pgi and pfkA genes and expressing glf and glk. Thus, metabolic engineering strategies are presented for improving yield and rate of reductive redox biocatalysis by partial cyclization of the PPP and by increasing glucose uptake, respectively.
Similar content being viewed by others
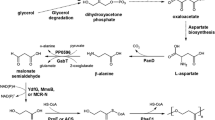

Introduction
Reductive whole-cell biotransformation is an important method for stereoselective industrial synthesis of chemical compounds. A variety of dehydrogenases catalyzes the enantio- and regioselective reduction of ketones and depends on nicotinamide adenine dinucleotide coenzymes (NADH or NADPH) for hydride transfer. Especially alcohol dehydrogenases are most interesting for the production of chiral alcohols because these enzymes show notable chemo-, regio-, and enantioselectivity (Goldberg et al. 2007). Efficient coenzyme recycling by recombinant enzymes converting the oxidized coenzyme back to its reduced form is essential for the productivity of reductive whole-cell biotransformations. In Escherichia coli, diverse strategies for cofactor regeneration have been applied: one-enzyme-coupled systems (Eguchi et al. 1992; Ernst et al. 2005; Seelbach et al. 1996; Wichmann and Vasic-Racki 2005) and approaches taking advantage of the metabolism of the cell (Walton and Stewart 2004; Chin et al. 2009; Fasan et al. 2011; Blank et al. 2008, 2010).
In a recent study with E. coli, we analyzed the NADPH-dependent reduction of the prochiral β-ketoester methyl acetoacetate (MAA) to the chiral hydroxy ester (R)-methyl 3-hydroxybutyrate (MHB) using glucose as substrate for the generation of NADPH (Siedler et al. 2011). The reduction is catalyzed by an R-specific alcohol dehydrogenase (ADH) from Lactobacillus brevis, and MHB serves as a building block of statins (Panke and Wubbolts 2005). Redirection of glucose catabolism from glycolysis to the pentose phosphate pathway (PPP) was accomplished by deletion of phosphoglucose isomerase (pgi) or the phosphofructokinase isoenzyme (pfkA, pfkB) genes resulting in a higher MHB to glucose yield (Siedler et al. 2011). Similar results were obtained in a parallel study analyzing the reduction of xylose to xylitol in E. coli with an NADPH-dependent xylose reductase from Candida boidinii (Chin and Cirino 2011). Based on the product/glucose yields obtained with the ΔpfkAΔpfkB mutant, a partial cyclization of the PPP was assumed because these yields were near the theoretical maximum yield of 6 mol reduced product per mole of glucose consumed (Kruger and von Schaewen 2003).
To further elucidate and verify this assumption, we performed 13C-based metabolic flux analyses with the ΔpfkA mutant in comparison to the Δpgi mutant, where a linear flux through the PPP is anticipated, as well as with the reference strain E. coli BL21(DE3). 13C-based metabolic flux analysis is a key technology for attaining an overview on the carbon fluxes within the cell. It also represents the only technique to date that allows the determination of the cofactor regeneration rate, which is a valuable parameter for estimating for the capacity of metabolism for redox biocatalysis (Blank et al. 2008).
Furthermore, we tested the influence of an alternative glucose uptake system on the performance of the reductive biotransformation. For this purpose, we introduced into the E. coli strains the glf and glk genes of Zymomonas mobilis ATCC29191, which encode the glucose facilitator (Glf) and glucokinase (Glk). Z. mobilis has a three to ten times higher glucose uptake rate than E. coli, which is due to the high velocity of the glucose facilitator Glf (Parker et al. 1995; Weisser et al. 1995). Overexpression of glf is feasible for increasing the glucose uptake rate of E. coli and was successfully used in combination with the cofactor regenerating system glucose dehydrogenase for production of α-pinene oxide (Schewe et al. 2008) and mannitol (Kaup et al. 2004, 2005).
Materials and methods
Bacterial strains and plasmids
Strains and plasmids used in this work are listed in Table 1. The mutant strains were constructed as described previously (Siedler et al. 2011). E coli strains were transformed by the method described by Hanahan (1983). Plasmids were selected by adding antibiotics to the medium at final concentrations of 100 μg ampicillin per milliliter (pBtac-Lbadh) and 50 μg kanamycin (pVWEx1, pVWEx1-glf-glk).
Media and growth conditions
Cells were cultivated in LB medium, 2xYT medium (16 g L−1 tryptone, 10 g L−1 yeast extract, 5 g L−1 sodium chloride) or M9 minimal medium (Sambrook and Russell 2001) containing 4 g L−1 glucose and supplemented with 1 mg mL−1 thiamine, 4 mg L−1 FeSO4 × 7 H2O, 1 mg L−1 MnSO4, 0.2 mg L−1 ZnSO4 × 7 H2O, 0.4 mg L−1 CoCl2, 0.1 mg L−1 CuCl2, 0.2 mg L−1 Na2MoO4 × 2 H2O, and 0.005 mg L−1 H3BO3. Solid media contained 1.5% (w/v) agar. Liquid cultures were routinely incubated in test tubes or 500-mL-baffled Erlenmeyer flasks overnight at 37 °C and 130 rpm. For stock cultures, 1 mL of overnight LB culture was gently mixed with 1 mL 30% (v/v) glycerol and stored at −80 °C.
13C-Labeling experiments
The first preculture of E. coli was performed in 5 mL of LB medium for 9 h at 37 °C and 130 rpm. The cells were then transferred to the second preculture containing 10 mL of M9 minimal medium and grown overnight at 37 °C and 130 rpm, followed by inoculation of 50 mL of M9 medium after a washing step with M9 medium without glucose to an OD600 of 0.05. In the main culture (50 mL M9 medium in a 500-mL Erlenmeyer flask) a mixture of 80% naturally labeled glucose and 20% [U-13C] glucose was used. Samples were taken to determine the growth rate and the glucose uptake rate. For 13C, metabolic flux analysis cells from 2-mL cultures were harvested during the exponential growth phase at an OD600 of ~0.7.
The cells were washed with cold 2.7% (w/v) NaCl solution; the pellets were resuspended in 6 M HCl and incubated for 24 h at 105 °C for hydrolysis of proteins. The samples were dried in a Speedvac (Eppendorf concentrator 5301) under a hood. The dried hydrolysates were derivatized for Gas chromatography and mass spectrometry (GC–MS) analysis using 50 μL N,N-dimethylformamide and 50 μL N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide. GC–MS analysis was carried out using a GC 3800, combined with an MS/MS 1200 unit (Varian Deutschland GmbH, Darmstadt, Germany). Fifteen detectable amino acids were separated on a FactorFour VF-5ms column (Varian Deutschland GmbH) at a constant flow rate of 1 mL helium per minute. The split ratio was 1:20, the injection volume was 1 μL, and the injector temperature was 250 °C. The temperature of the GC oven was kept constant for 2 min at 150 °C and was afterwards increased to 250 °C with a slope of 3 °C min−1. The temperature of the transfer line and the source was 280 and 250 °C, respectively. Ionization was performed by electron impact ionization at −70 eV. For enhanced detection, a selected ion monitoring frame was defined for every amino acid (Wittmann 2007) GC–MS raw data were analyzed using the Workstation MS Data Review (Varian Deutschland GmbH) to check for detector overload (Heyland et al. 2009).
13C-constrained metabolic flux analysis
The stoichiometric model, implemented in FiatFlux (Zamboni et al. 2005b) for 13C-constrained metabolic flux analysis comprises the major pathways of E. coli central carbon metabolism (Fischer et al. 2004). To calculate intracellular fluxes, the stoichiometric model was constrained with three extracellular fluxes (growth rate, glucose uptake rate, and acetate formation rate) and five intracellular flux ratios (fraction of oxaloacetate originating from phosphoenolpyruvate, fraction of oxaloacetate derived through anaplerosis, fraction of phosphoenolpyruvate originating from oxaloacetate, and upper and lower bounds of pyruvate derived through malic enzyme). Error minimization during flux calculation using the determined equation system was carried out as described previously (Fischer et al. 2004). The method for the determination of flux ratios is described in detail by Nanchen and coworkers (Nanchen et al. 2007), while the 13C-constrained flux analysis is described by Zamboni and coworkers (Zamboni et al. 2009).
Whole-cell biotransformation
For cultivation of the different recombinant E. coli, BL21(DE3) strains carrying the plasmids pBtac-Lbadh and pVWEx1 or pVWEx1-glf-glk, a single colony of each strain, was inoculated into 5 mL of 2xYT medium containing the appropriate antibiotics and grown overnight at 37 °C and 170 rpm. These pre-cultures were used for inoculation of the main cultures to an OD600 of 0.05. Main cultures were grown in 100 mL of 2xYT medium in the presence of the appropriate antibiotics at 37 °C and 130 rpm to an OD600 of 0.3, induced with 1 mM of IPTG, incubated at 37 °C and 130 rpm for another 3 h and harvested at an OD600 between 2 and 4.5, which had been determined as the optimal cell density for subsequent whole-cell biotransformation.
The cells were harvested by centrifugation (4,000×g, 7 min) and resuspended in reaction buffer containing 278 mM glucose, 2 mM MgSO4, and 250 mM potassium phosphate buffer, pH 6.5, to a cell density of 3 g cell dry weight (cdw) per liter. The reaction started by adding 60 mM of MAA. Biotransformations were performed at 37 °C and 130 rpm to prevent cell sedimentation. Specific productivities (millimole MHB per hour per gram cdw) were determined by taking samples at 15-min time intervals over a period of 2 h. MHB and glucose concentrations of the samples were determined (see below). Specific productivities were calculated by dividing the slope of graphs showing MHB concentration vs. time by the cell dry weight.
Analysis of substrates and products
Methyl acetoacetate (MAA), (R)-methyl 3-hydroxybutyrate (MHB), glucose and extracellular metabolites were analyzed by HPLC as described previously (Siedler et al. 2011). Glucose concentrations were also determined enzymatically by using the glucose Gluc-DH FS* Kit according to the instructions of the manufacturer (DiaSys, Holzheim, Germany).
Results
Growth behavior of reference and mutant strains
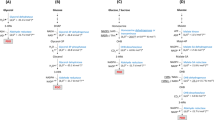
The growth kinetics of the reference and deletion strains were determined in M9 minimal medium (Fig. 1). The reference strain E. coli BL21(DE3) had the highest growth rate of 0.60 h−1 followed by the ΔpfkA mutant and the Δpgi mutant with 0.18 and 0.11 h−1, respectively (Table 2). The specific glucose uptake rate was very high for the reference strain (11.3 mmol h−1 g −1cdw ) but in the range of previously reported values (Toya et al. 2010) and significantly lower for the ΔpfkA mutant (4.0 mmol h−1 g −1cdw ) and the Δpgi mutant (2.7 mmol h−1 g −1cdw ). We chose the ΔpfkA mutant instead of the ΔpfkAΔpfkB mutant for flux analysis because of the very slow growth of the latter strain in glucose minimal media (Table 2).

a Growth of E. coli BL21(DE3) (black triangle) and the mutants ΔpfkA (white triangle) and Δpgi (gray triangle). b Glucose uptake of E. coli BL21(DE3) (black square) and the mutants ΔpfkA (white square) and Δpgi (gray square)
In order to show cyclization of the PPP and to gain more insight into the metabolic network upon genetic perturbation of glycolysis, we measured the carbon fluxes of the reference strain and of the deletion strains Δpgi and ΔpfkA during growth in minimal medium with glucose as the sole carbon source (Fig. 2). Samples were taken in the early exponential growth phase at an OD600 of ~0.7 where pseudo steady state conditions can be assumed. The reference strain showed similar flux distributions as reported previously for a batch culture with exception of a higher flux through the tricarboxylic acid (TCA) cycle and a much lower acetate formation (Toya et al. 2010). These differences might be due to the different origins of the E. coli B strain BL21(DE3) and the E. coli K12 strain BW25113 used by Toya and coworkers. The Δpgi mutant of BL21(DE3) showed a linear flux through the PPP, similar to the BW25113-derived Δpgi mutant, but again a twofold higher flux through the TCA cycle. The ΔpfkA mutant of BL21(DE3) showed a partial cyclization of the PPP with a negative net flux through the reaction from glucose 6-phosphate to fructose 6-phosphate catalyzed by phosphoglucose isomerase. This was consistent with the estimated carbon flux-based on the molar product per glucose yield achieved in the biotransformations reported previously (Chin and Cirino 2011). According to our flux analysis, the ΔpfkA mutant possessed about 30% residual phosphofructokinase activity. This value is close to the one obtained by in vitro enzyme activity measurements with cell extracts, which revealed 20% residual phosphofructokinase activity in this mutant (Siedler et al. 2011). The residual activity is due to the PfkII isoenzyme.

Results of 13C-based metabolic flux analysis of E. coli BL21(DE3) (upper number) and mutant strains Δpgi (middle number) and ΔpfkA (lower number) during exponential growth on glucose. In vivo carbon fluxes in central metabolism are shown as millimole per hour per gram cdw. Relative to the glucose uptake rate, the confidence intervals were 20% for the NADPH balance, below 15% for TCA cycle and malic enzyme, and less than 5% for all other fluxes. Arrowheads indicate the direction of a given flux
Additionally, we calculated the NADH regeneration rates and the fluxes through the soluble transhydrogenase for an estimation of the reduced cofactor availability in the cells (Table 3). The NADH regeneration rates relative to the glucose uptake rates of all strains were similar, in contrast to a significantly higher flux through the soluble transhydrogenase UdhA in the ΔpfkA mutant (Table 3).
Improvement of glucose uptake in whole-cell biotransformations
The 13C metabolic flux analysis had indicated a very low flux through the reaction catalyzed by pyruvate kinase in the Δpgi and especially the ΔpfkA mutant (Fig. 2), indicating that glucose uptake could be limited by PEP availability. This was also supported by the much lower specific glucose uptake rates of these two mutants (Table 2). Therefore, we tried to increase the glucose uptake rate and possibly also the biotransformation rate by introduction of an additional glucose uptake system. The glucose facilitator Glf from Z. mobilis, a transporter with a high velocity but a low affinity (Parker et al. 1995; Weisser et al. 1995), and the glucose kinase Glk of the same organism was chosen as an alternative pathway for increasing the rate of glucose uptake. As in our previous studies, the NADPH-dependent reduction of MAA to MHB by the (R)-specific alcohol dehydrogenase of Lactobacillus brevis was used as a model reaction for biotransformation, and the corresponding adh gene was overexpressed in all strains.
Overexpression of glf and glk using plasmid pVWEx1-glf-glk in the reference strain SS01 (E. coli BL21(DE3) containing pBTac-Lbadh) led to a 46% increased specific glucose uptake rate (Table 4), confirming the functional expression of the heterologous genes. However, the higher glucose uptake rate did not result in a higher biotransformation rate compared to the control strain SSO1 with plasmid pVWEx1. The molar yield of MHB per glucose was even decreased by 34% in strain SS01/pVWEx1-glf-glk (Table 4). Expression of glf and glk in strains SS02 (containing the pgi deletion) and SS03 (containing the pfkA deletion) led to 30% and 41% increased glucose uptake rates, respectively, and also to 15% and 20% improved MHB production rates, compared to the corresponding strains with the empty vector pVWEx1. The ratio of MHB produced per glucose consumed decreased also in these strains by 11% (Δpgi mutant) and 6% (ΔpfkA mutant) compared to the corresponding reference strains. This decrease could be due to the overexpression of glf and glk, leading to a high metabolic burden of heterologous protein production (Schweder et al. 2002; Dong et al. 1995; Bhattacharya and Dubey 1995). In the case of strain SS04 (deletion of pfkA and pfkB), overexpression of glf and glk led to a 40% increased glucose uptake rate, whereas the MHB production rate was slightly decreased by 4% and the molar yield by 31% compared to the reference strain. The presence of the vector pVWEx1 showed no significant influence in strains SS01, SS02, and SS03 compared to the strains without this plasmid, but caused a much lower specific MHB production rate by strain SS04 compared to SS04 without pVWEx1.
Discussion
To improve reductive biotransformation we engineered the metabolic network of E. coli. Optimization of the yield was achieved by deleting phosphofructokinase genes resulting in a partial cyclization of the PPP. An increase of the glucose uptake rate by overexpression of the glucose facilitator gene glf, and the glucokinase gene glk from Zymomonas mobilis resulted in a higher specific biotransformation rate in the strains SS02 (pgi deletion) and SS03 (pfkA deletion).
Partial cyclization of the PPP in strain SS03 was confirmed by 13C metabolic flux analysis of cells grown in glucose minimal medium, which revealed a negative net flux from glucose 6-phosphate to fructose 6-phosphate catalyzed by phosphoglucose isomerase. Under resting cell conditions, this partial cyclization resulted in a significant increase of the whole-cell biotransformation yield. A yield of 4.7 molMHB mol −1Glucose was attained with strain SS03, which contained residual phosphofructokinase activity originating from pfkB. Although not shown by 13C flux analysis, cyclic flux through the PPP should be even more intense in strain SS04 (pfkApfkB deletion) because the yield of strain SS04 with 5.5 molMHB mol −1Glucose almost reached the theoretical maximum yield of 6 molNADPH mol −1Glucose (Kruger and von Schaewen 2003). Strain SS02 displayed a somewhat higher yield (3.7 molMHB mol −1Glucose ) than theoretically expected for reasons discussed recently (Siedler et al. 2011).
The Δpgi and ΔpfkA mutant strains showed strongly reduced glucose uptake rates, in agreement with previous studies (Sauer et al. 2004; Nanchen et al. 2007). Several reasons for the lower glucose uptake rate have already been discussed in literature. The Δpgi mutant has a high glucose 6-phosphate pool, which was reported to destabilize the ptsG mRNA and therefore decreases the glucose uptake capacity of the cell (Morita et al. 2003). For the ΔpfkA mutant, a second assumption was reported by Roehl and Vinopal (1976) according to which the reduced PEP concentration in this mutant decreases the glucose uptake rate because PEP is essential for glucose uptake by the phosphotransferase system. This assumption is consistent with the carbon fluxes through the pyruvate kinase reaction determined here, which were lower in the Δpgi mutant and nearly absent in the ΔpfkA mutant (Fig. 2).
In the SS01 strain, an increased glucose uptake rate did not result in higher rate of MAA reduction to MHB, indicating that another factor was limiting the rate of biotransformation, presumably NADPH availability (see below). In strains SS02 and SS03, glucose uptake limited the specific biotransformation rate, as shown by the increased MHB production rate upon overexpression of glf and glk.
For whole-cell biotransformation several limitations are possible, such as, enzyme activity, substrate import and product export, product toxicity, and cofactor regeneration rate, but most of them can be excluded in the model system used here. The alcohol dehydrogenase showed in vitro activities of 17 to 28 U mg−1 in cell extracts (Siedler et al. 2011), which is comparable to a rate of at least 418 mmol h−1 g −1cdw in vivo assuming that 41% of the cell dry weight is made up of soluble proteins (Zamboni et al. 2005a; Wittmann et al. 2007). This value is 40 times higher than the observed specific biotransformation rate and consequently, ADH activity is not limiting. Substrate and product transport can also be ruled out as limiting factors, because biotransformation rates of 33 mmolMHB h−1 g −1cdw were previously achieved using a substrate coupled biotransformation strategy with ADH catalyzing both, the reduction of MAA and the regeneration of NADPH by oxidation of 2-propanol (Schroer et al. 2009). With a formate dehydrogenase-coupled NADPH regeneration system, a value of 12 mmolMHB h−1 g −1cdw has been reported (Ernst et al. 2005). For the conversion of MAA to MHB, it was shown that there is no substrate and product inhibition, and neither substrate nor product are toxic to the cells under the chosen conditions (Tan 2006). Taken together, the biotransformation system used in this work is applicable for the detection of cofactor regeneration capabilities because this seems to be the limiting step.
Calculation of the NADH regeneration rate and transhydrogenase activity from 13C flux analysis in growing cells revealed differences in the analyzed strains (Fig. 2). The relative fluxes through the soluble transhydrogenase UdhA converting NADPH to NADH were 2.5 and 4.1 times higher in the Δpgi mutant and the ΔpfkA mutant compared to the reference strain, respectively (Table 3). The yield of the biotransformation increased 1.3-fold in the SS02 strain and 1.6-fold in the SS03 strain compared to the SS01 strain, which indicated that under resting cell conditions ADH was competitive to UdhA with respect to NADPH utilization. Hence, deletion of UdhA would probably result in an increased yield, as was already demonstrated with another biotransformation system (Chin and Cirino 2011).
Having increased product yield and production rate by different strategies, future studies aim to combine these and additional strategies for further optimization of reductive whole-cell redox biocatalysis.
References
Bhattacharya SK, Dubey AK (1995) Metabolic burden as reflected by maintenance coefficient of recombinant Escherichia coli overexpressing target gene. Biotechnol Lett 17:1155–1160
Blank LM, Ebert BE, Bühler B, Schmid A (2008) Metabolic capacity estimation of Escherichia coli as a platform for redox biocatalysis: constraint-based modeling and experimental verification. Biotechnol Bioeng 100(6):1050–1065
Blank LM, Ebert BE, Bühler K, Bühler B (2010) Redox biocatalysis and metabolism: molecular mechanisms and metabolic network analysis. Antioxid Redox Signal 13(3):349–394
Chin JW, Cirino PC (2011) Improved NADPH supply for xylitol production by engineered Escherichia coli with glycolytic mutations. Biotechnol Prog 27(2):333–341
Chin JW, Khankal R, Monroe CA, Maranas CD, Cirino PC (2009) Analysis of NADPH supply during xylitol production by engineered Escherichia coli. Biotechnol Bioeng 102(1):209–220
Dong H, Nilsson L, Kurland CG (1995) Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. J Bacteriol 177(6):1497–1504
Eguchi T, Kuge Y, Inoue K, Yoshikawa N, Mochida K, Uwajima T (1992) NADPH regeneration by glucose dehydrogenase from Gluconobacter scleroides for l-leucovorin synthesis. Biosci Biotechnol Biochem 56(5):701–703
Ernst M, Kaup B, Müller M, Bringer-Meyer S, Sahm H (2005) Enantioselective reduction of carbonyl compounds by whole-cell biotransformation, combining a formate dehydrogenase and a (R)-specific alcohol dehydrogenase. Appl Microbiol Biotechnol 66(6):629–634
Fasan R, Crook NC, Peters MW, Meinhold P, Buelter T, Landwehr M, Cirino PC, Arnold FH (2011) Improved product-per-glucose yields in P450-dependent propane biotransformations using engineered Escherichia coli. Biotechnol Bioeng 108(3):500–510
Fischer E, Zamboni N, Sauer U (2004) High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem 325(2):308–316
Goldberg K, Schroer K, Lütz S, Liese A (2007) Biocatalytic ketone reduction-a powerful tool for the production of chiral alcohols-part II: whole-cell reductions. Appl Microbiol Biotechnol 76(2):249–255
Hanahan D (1983) Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166(4):557–580
Heyland J, Fu J, Blank LM (2009) Correlation between TCA cycle flux and glucose uptake rate during respiro-fermentative growth of Saccharomyces cerevisiae. Microbiology 155(Pt 12):3827–3837
Kaup B, Bringer-Meyer S, Sahm H (2004) Metabolic engineering of Escherichia coli: construction of an efficient biocatalyst for D-mannitol formation in a whole-cell biotransformation. Appl Microbiol Biotechnol 64(3):333–339
Kaup B, Bringer-Meyer S, Sahm H (2005) D-Mannitol formation from D-glucose in a whole-cell biotransformation with recombinant Escherichia coli. Appl Microbiol Biotechnol 69(4):397–403
Kruger NJ, von Schaewen A (2003) The oxidative pentose phosphate pathway: structure and organisation. Curr Opin Plant Biol 6(3):236–246
Morita T, El-Kazzaz W, Tanaka Y, Inada T, Aiba H (2003) Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J Biol Chem 278(18):15608–15614
Nanchen A, Fuhrer T, Sauer U (2007) Determination of metabolic flux ratios from 13 C-experiments and gas chromatography-mass spectrometry data: protocol and principles. Methods Mol Biol 358:177–197
Panke S, Wubbolts M (2005) Advances in biocatalytic synthesis of pharmaceutical intermediates. Curr Opin Chem Biol 9(2):188–194
Parker C, Barnell WO, Snoep JL, Ingram LO, Conway T (1995) Characterization of the Zymomonas mobilis glucose facilitator gene product (glf) in recombinant Escherichia coli: examination of transport mechanism, kinetics and the role of glucokinase in glucose transport. Mol Microbiol 15(5):795–802
Peters-Wendisch PG, Schiel B, Wendisch VF, Katsoulidis E, Mockel B, Sahm H, Eikmanns BJ (2001) Pyruvate carboxylase is a major bottleneck for glutamate and lysine production by Corynebacterium glutamicum. J Mol Microbiol Biotechnol 3(2):295–300
Roehl RA, Vinopal RT (1976) Lack of glucose phosphotransferase function in phosphofructokinase mutants of Escherichia coli. J Bacteriol 126(2):852–860
Sambrook J, Russell DW (eds) (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spting Harbor
Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E (2004) The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem 279(8):6613–6619
Schewe H, Kaup BA, Schrader J (2008) Improvement of P450(BM-3) whole-cell biocatalysis by integrating heterologous cofactor regeneration combining glucose facilitator and dehydrogenase in E. coli. Appl Microbiol Biotechnol 78(1):55–65
Schroer K, Zelic B, Oldiges M, Lütz S (2009) Metabolomics for biotransformations: Intracellular redox cofactor analysis and enzyme kinetics offer insight into whole cell processes. Biotechnol Bioeng 104(2):251–260
Schweder T, Lin HY, Jürgen B, Breitenstein A, Riemschneider S, Khalameyzer V, Gupta A, Büttner K, Neubauer P (2002) Role of the general stress response during strong overexpression of a heterologous gene in Escherichia coli. Appl Microbiol Biotechnol 58(3):330–337
Seelbach K, Riebel B, Hummel W, Kula MR, Tishkov VI, Egorov AM, Wandrey C, Kragl U (1996) A novel, efficient regenerating method of NADPH using a new formate dehydrogenase. Tetrahedron Lett 37:1377–1380
Siedler S, Bringer S, Bott M (2011) Increased NADPH availability in Escherichia coli: improvement of the product per glucose ratio in reductive whole-cell biotransformation. Appl Microbiol Biotechnol. doi:10.1007/s00253-011-3374-4
Tan I (2006) Applications of whole cell biotransformations for the production of chiral alcohols. Rheinische Friedrich-Wilhelms University of Bonn, Bonn
Toya Y, Ishii N, Nakahigashi K, Hirasawa T, Soga T, Tomita M, Shimizu K (2010) 13C-metabolic flux analysis for batch culture of Escherichia coli and its pyk and pgi gene knockout mutants based on mass isotopomer distribution of intracellular metabolites. Biotechnol Prog 26(4):975–992
Walton AZ, Stewart JD (2004) Understanding and improving NADPH-dependent reactions by nongrowing Escherichia coli cells. Biotechnol Prog 20(2):403–411
Weisser P, Krämer R, Sahm H, Sprenger GA (1995) Functional expression of the glucose transporter of Zymomonas mobilis leads to restoration of glucose and fructose uptake in Escherichia coli mutants and provides evidence for its facilitator action. J Bacteriol 177(11):3351–3354
Wichmann R, Vasic-Racki D (2005) Cofactor regeneration at the lab scale. Adv Biochem Eng Biotechnol 92:225–260
Wittmann C (2007) Fluxome analysis using GC-MS. Microb Cell Fact 6:6
Wittmann C, Weber J, Betiku E, Kromer J, Bohm D, Rinas U (2007) Response of fluxome and metabolome to temperature-induced recombinant protein synthesis in Escherichia coli. J Biotechnol 132(4):375–384
Zamboni N, Fischer E, Muffler A, Wyss M, Hohmann HP, Sauer U (2005a) Transient expression and flux changes during a shift from high to low riboflavin production in continuous cultures of Bacillus subtilis. Biotechnol Bioeng 89(2):219–232
Zamboni N, Fischer E, Sauer U (2005b) FiatFlux—a software for metabolic flux analysis from 13C-glucose experiments. BMC Bioinformatics 6:209
Zamboni N, Fendt SM, Ruhl M, Sauer U (2009) 13C-based metabolic flux analysis. Nat Protoc 4(6):878–892
Acknowledgments
This work was supported by the Ministry of Innovation, Science, Research and Technology of North Rhine-Westphalia (BioNRW, Technology Platform Biocatalysis, RedoxCell support code: W0805wb001b).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Siedler, S., Bringer, S., Blank, L.M. et al. Engineering yield and rate of reductive biotransformation in Escherichia coli by partial cyclization of the pentose phosphate pathway and PTS-independent glucose transport. Appl Microbiol Biotechnol 93, 1459–1467 (2012). https://doi.org/10.1007/s00253-011-3626-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-011-3626-3