
Abstract
In the last decade, a new gene family encoding non-rearranging receptors, called novel immune-type receptors (NITRs), has been discovered in teleost fish. NITRs belong to the immunoglobulin superfamily and represent an extraordinarily divergent and rapidly evolving gene complex. Genomic analysis of a region spanning 270 kb led to the discovery of a NITR gene cluster in the European sea bass (Dicentrarchus labrax). In total, 27 NITR genes and three putative pseudogenes, organised in a tandemly arrayed cluster, were identified. Sea bass NITR genes maintain the three major genomic organisations that appear to be essentially conserved among fish species along with new features presumably involving processes of intron loss, exon deletion and acquisition of new exons. Comparative and evolutionary analyses suggest that these receptors have evolved following a “birth-and-death” model of gene evolution in which duplication events together with lineage-specific gain and loss of individual members contributed to the rapid diversification of individual gene families. In this study, we demonstrate that species-specific gene expansions provide the raw material for diversifying, positive Darwinian selection favouring the evolution of a highly diverse array of molecules.
Similar content being viewed by others

Introduction
Increasing attention is being directed to the role of innate processes within the immune system, the variety and diversity of innate immune receptors, their interaction with other surface molecules and the mechanisms employed for transduction of intracellular signals. Several clusters of innate immune receptors have been identified in mammals. In primates, particular attention has been given to receptors belonging to the immunoglobulin superfamily; the killer Ig-like receptors (KIR; Martinez-Borra and Khakoo 2008), the Ig-like transcripts (ILT; Samaridis and Colonna 1997; leukocyte inhibitory receptors and monocyte inhibitory receptors), the leukocyte-associated inhibitory receptors (Meyaard et al. 1997) and the Fc receptors. Interestingly, all these genes, which are structurally and functionally related, are encoded in the same region of the human chromosome 19q13.4, termed the leukocyte receptor complex (LRC; Torkar et al. 1998), and also in non-human primates where they show a tandem arrangement (Canavez et al. 2001). KIR receptors, members of the natural killer complex (NKC), have been proposed to interact with MHC class I molecules, enabling NK cells to discern target cells exposing “missing self” signals (Yokoyama and Plougastel 2003). These receptors are type I transmembrane proteins characterised by the presence of a variable number of extracellular Ig domains of either the C2-type (I) or the novel V-type (V). The Ig domains are followed by a stem region, a transmembrane region (TM) and a cytoplasmic tail (Cyt). Depending on the cytoplasmic tail, they can be subdivided into inhibitory and activating receptors. In general, inhibitory KIRs possess long cytoplasmic tails containing one or two immunoreceptor tyrosine-based inhibitory motifs (ITIM), whereas short-tailed KIRs generate activating signals. In the last decade, two gene families encoding non-rearranging receptors with a similar molecular structure to that of KIRs have been discovered in teleost fish, the novel immune-type receptors (NITRs) and the novel immunoglobulin-like transcripts (NILTs). So far, two and four NILTs have been isolated from the common carp (Stet et al. 2005) and the rainbow trout (Kock and Fischer 2008; Østergaard et al. 2008), respectively. NILTs possess one or two V-type Ig domains followed by a transmembrane domain and a cytoplasmic tail containing one immunoreceptor tyrosine-based activation motif (ITAM) and/or a variable number of ITIM domains.
NITR genes have been identified in several fish species. Twenty-six individual genes, all located on a single genomic region spanning ?113 kb, were first characterised in the Southern pufferfish (Strong et al. 1999), then followed the isolation and description of four NITRs in the rainbow trout (Yoder et al. 2002), 18 individual genes in the channel catfish (Hawke et al. 2001; Evenhuis et al. 2007) and one transcript in the Japanese flounder (Piyaviriyakul et al. 2007). NITRs are best characterised in the zebrafish in which 12 families were found on chromosome (chr.) 14 and two on chr. 7 (Yoder et al. 2001, 2004) and in medaka with the identification of 24 families located in three gene clusters, respectively, on chromosomes 10, 18 and 21 (Desai et al. 2008). Transcripts encoding a NITR protein have also been found in the Atlantic salmon, Atlantic cod, Atlantic halibut, lake whitefish and stickleback (Yoder 2009). NITRs appear to represent a large and highly diversified gene complex, with several forms identified according to: (1) number of extracellular Ig domains (V-type and I-type), (2) number of exons encoding the ectodomains, (3) presence and location of joining (J) or J-like sequences, (4) presence of ITIM motifs in the cytoplasmic tail and (5) presence of charged residues in the transmembrane region. NITRs have been proposed to be functional homologs of mammalian KIRs, as both NITRs and KIRs appear to stimulate the same signalling pathways (Yoder 2009). In zebrafish, activating NITRs have been shown to redirect NK cell-mediated cytolysis by interaction with the fish ortholog of DAP12 (Yoder et al. 2004), whilst an inhibitory activity has been proposed for ITIM and ITIM-like domains, exerted through recruitment of the tyrosine phosphatases SHP-1 and SHP-2 and/or the inositol phosphatase SHIP-1, which transduces negative signals (Bléry et al. 2000; Yoder et al. 2007; Piyaviriyakul et al. 2007). Moreover, the crystal structure of NITR Ig domains provides evidence that binding specificity for the allogeneic determinant resides in the V region CDR1-analogous loop and demonstrates their potential interaction with a polymorphic set of ligands (Cannon et al. 2008), like NK cell receptors.
Even though NITR genes are structurally similar to certain members of NKC, their origin remains to be elucidated. Comparative genomic analysis between the NITR gene clusters and mammalian LRC showed only a weakly conserved synteny, and it cannot be excluded that the presence of NITRs may be restricted to teleost fish (Yoder 2009). Data collected so far demonstrate that NITRs represent an extraordinarily divergent and rapidly evolving gene family. Different NITR profiles and a remarkable diversity of gene content were found across teleost species, suggesting that independent, lineage-specific tandem gene duplications and family expansions are the most frequent mode for the NITR gene complex evolution (Litman et al. 2001; Yoder 2009). The functional significance of such lineage-specific gene expansions remains to be further investigated.
In this paper, we describe the identification and characterisation of 27 NITRs in the European sea bass (Dicentrarchus labrax), all encoded in a single genomic region spanning 270 kb. Classified into at least 18 families, sea bass NITRs represent a highly diversified gene complex generated by repeated tandem duplication events followed by rapid diversification. The resolution of this gene cluster together with the previous genomic characterisation of NITRs in zebrafish, medaka and Southern pufferfish allowed an exhaustive evolutionary analysis as well as the examination of shared synteny.
The European sea bass is a marine teleost fish of great importance for fisheries and aquaculture, being an excellent food fish with high commercial value. The great importance of D. labrax for marine aquaculture has fuelled an increasing number of studies in many different areas such as immunology, endocrinology, bone morphology and muscle physiology, whilst genomic resources for this species have been constantly improving, leading to a whole genome sequencing project which is now well advanced (R. Reinhardt, unpublished data). In particular, the D. labrax immune system is one of the best characterised among teleost species (Scapigliati et al. 2002; Randelli et al. 2009), providing a nice model for immunological studies, which is complementary to the best fish models such as the channel catfish and zebrafish. These two are freshwater fish belonging to Ostariophysi, a less derived group, whilst D. labrax is a marine fish representative of the highly derived Perciform order, which is the most species-rich vertebrate group with over 20,000 species (Nelson 2006).
Materials and methods
In silico identification of sea bass NITRs
A first bacterial artificial chromosome (BAC) putatively containing sea bass NITRs was found through Blast analysis of sequenced BAC-ends of a sea bass BAC library (Kuhl et al., in preparation). After complete shotgun sequencing of the first BAC (bassbac-18k1), a second BAC (bassbac-79e10) was sequenced, which potentially contained a further fragment of the candidate genomic region based on Blast analysis.
The DNA of identified BAC clones was isolated by alkaline lysis, and subsequently remaining Escherichia coli DNA was removed by ATP-dependent exonuclease digestion. Purified BAC-DNA was sheared by ultrasonic sound, and fragment sizes of 1–4 kb were selected for end-polishing with T4 DNA polymerase and DNA polymerase I (Klenow). Fragments were ligated with T4 DNA ligase into the SmaI restriction site of puC19 sequencing vector. Competent E. coli DH10B cells were transformed by electroporation. For each BAC, a library with approximately tenfold coverage was constructed, and plasmid DNA was purified for sequencing with ABI BigDye v3.1 Terminator chemistry on ABI3730xl (Applied Biosystems Inc., Foster City, CA, USA) capillary sequencers. Raw sequences were processed by PHRED (Ewing and Green 1998), and removal of vector backbone or low-quality sequence was done by LUCY (Chou and Holmes 2001). The remaining sequences were screened for E. coli or BAC vector contamination by megablast. The BAC inserts were assembled using PHRAP (available from Phil Green, University of Washington; www.phrap.org).
In order to identify NITR genes in the sea bass genomic contig, all the publicly available NITR protein sequences were used as queries for Blast analyses (Wolfsberg and Madden 2001) using the tblastn option. The APOLLO software (Lewis et al. 2002) was used to visualise the organisation of the analysed genomic region and to manually annotate NITR genes structures based on all the available supporting evidence. Signal peptide/leader sequences were predicted using SignalP 3.0 server (http://www.cbs.dtu.dk/services/SignalP/), whilst TMHMM v2.0 server (http://www.cbs.dtu.dk/services/TMHMM/) was used to search for transmembrane domains.
Families of sea bass NITRs were defined using the MatGat programme (MATrix global alignment tool; Campanella et al. 2003) based on the criterion of 70% shared identity within the peptide sequence of the V domain.
Tissue collection and RNA extraction
Four juvenile sea bass individuals (17–20 g) were collected from the experimental aquaria of the Istituto Zooprofilattico Sperimentale delle Venezie (Padova, Italy) and sacrificed using an excess of anaesthetic. Eight different tissues/organs (gill, spleen, liver, intestine, skeletal muscle, skin, head kidney and whole blood) were collected from each animal and stored in RNALater® (Ambion, Austin TX, USA) at 4°C for 24 h followed by long-term storage at ?20°C. Total RNA was extracted using an RNAeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s specifications. The quality of the RNA was checked by gel electrophoresis on a 1% agarose gel containing SYBR Safe™ DNA gel stain ×10,000 (Invitrogen™, Carlsbad, CA, USA).
Amplification of NITR cDNA segments and full-length transcripts
In order to validate exon–intron boundaries and transmembrane predictions and to search for short sequence motifs, cDNA amplification and sequencing of all predicted NITR genes in the cluster was carried out using RNA extracted from D. labrax head kidney. One microgram of total RNA was reverse-transcribed to cDNA using Superscript II (Invitrogen™). Primer pairs for all the sea bass NITR genes were designed based on the in silico predicted gene sequence. Whenever possible, forward and reverse primers included the whole putative coding region of the gene. One microlitre of diluted (1:10) cDNA was used as template in PCR. Cycling conditions were: initial incubation at 94°C for 2 min followed by 45 cycles at 94°C for 45 s, 60°C for 30 s and 72°C for 45 s. A final extension step at 72°C for 5 min was added at the end of the last cycle. For each NITR gene, annealing temperature was set according to the predicted melting temperature of primers. Both 3? and 5? rapid amplification of cDNA ends reactions were also carried out for five transcripts.
Evolutionary analyses
Evolutionary analyses were performed to determine patterns of divergence of the NITR genes in D. labrax as well as to define putative orthology between NITR genes in different teleost species. All published protein sequences of NITR V domains were aligned using ClustalW (Thompson et al. 1994), applying default settings, whereas CodonAlign v2.0 was used to carry out the nucleotide alignment of V and I coding regions. NITR sequences described from medaka, zebrafish, Southern pufferfish, channel catfish, rainbow trout, Japanese flounder and green-spotted pufferfish were included in the alignment. It should be noted that sea bass NITRs, as well as other teleost ones, are named accordingly to the order that they were identified in each species. Thus, NITRs with the same name in different fish species are generally not orthologous.
Phylogenetic trees were inferred using the maximum likelihood (ML) method (Felsenstein 2004). For amino acid sequences, the most appropriate model of protein evolution was selected using the Akaike information criterion as implemented in ProtTest v1.4 (Abascal et al. 2005). The ML analyses were performed with the Phyml 2.4.4 programme (Guindon and Gascuel 2003) by applying the JTT+I+G+F evolutionary model; non-parametric bootstrap resampling (500 replicates) was also performed to evaluate the robustness of tree topology. For nucleotide sequences, ML analysis was performed by applying the GTR+I+G model. The TREEFINDER programme (Jobb et al. 2004) was then used in order to evaluate alternative phylogenetic hypotheses.
Tests for positive selection were carried out by comparing the number of non-synonymous substitutions per non-synonymous site (dN) with the number of synonymous substitutions per synonymous site (dS) in a ML framework. A ratio of dN/dS (?) greater than 1 is usually taken as evidence of selection following different methodological approaches. First, a series of nested models as implemented in the software PAML ver. 4 (Yang 2007) were evaluated, comparing “neutral” models, which restrict ? to values lower or equal to 1 (models M1, M7 and M8a) with models allowing for positive selection (? greater than 1 for a class of sites; models M2 and M8). Comparison of nested models was carried out using a likelihood ratio test (LRT). All analyses were performed twice starting from different initial values of ? to check for convergence in ML parameter estimates. Adopted models for codon frequencies were F3×4 and F61. Virtually identical results were obtained using both codon frequency models; thus, only data from the simplest one (F3×4) are reported. Amino acids under selection for model M8 were identified using a Bayes empirical Bayes approach (BEB) (Yang et al. 2005). BEB probabilities higher than 0.95 were considered significant. Two additional likelihood methods, incorporating variation in the rate of synonymous substitution, random effects likelihood (REL) and fixed effects likelihood (FEL; Kosakovsky Pond and Frost 2005) were implemented at the DATAMONKEY web server (Pond and Frost 2005). Finally, a codon model (MEC), which takes into account the physicochemical properties of amino acids when estimating non-synonymous substitution rates (Doron-Faigenboim and Pupko 2006), was implemented at the web server Selecton (Stern et al. 2007).
Comparative genomic analysis
Three proteins flanking the sea bass NITR gene cluster, RBM4 (RNA binding motif 4), TRAF2-binding protein (TNF receptor-associated factor 2) and FXR2 (fragile X mental retardation gene 2), were used to investigate the syntenic relationship between D. labrax and the other teleost species. Putative orthologs of these sea bass genes were found and their location identified with TBLASTN searches against the Danio rerio, Gasterosteus aculeatus and Oryzias latipes genomes. Latest genome releases of the Ensembl web site (http://www.ensembl.org/index.html) were used to achieve this task.
Comparative modelling of NITR V domain
Theoretical models of the Dl28 family V domains were generated with Modeller 9v4 (http://www.salilab.org/modeller/), with symmetry constraints applied to build symmetry-related domain dimers. The comparative modelling approach implemented in Modeller was based on the satisfaction of spatial restraints obtained from the alignment of the target sequence to one or more model structures (Sali and Blundell 1993). The recently solved Ictalurus punctatus NITR10 and NITR11 (Cannon et al. 2008) were selected as templates according to the highest sequence similarity. Firstly, sea bass sequences were used to perform a FASTA search on the Protein Data Bank (http://www.rcsb.org/) for each NITR domain. Next, the ClustalW server (http://www.ebi.ac.uk/Tools/clustalw2/index.html) was used to align V domains for each target/template combination. The model-default routine of Modeller was then used to generate five protein models for each sea bass sequence. Best models were selected based on their stereochemical properties.
Expression of NITR mRNAs in D. labrax
NITR mRNA expression, in the eight tissues listed above, was examined by real-time polymerase chain reaction (RT-PCR) for four individual fishes. Total RNA extraction and RT-PCR were performed as described earlier. To examine expression of NITRs, ?-actin was amplified as positive control. Amplicons were then resolved by gel electrophoresis in 1.8% agarose containing SYBR Safe™ DNA gel stain ×10,000 (Invitrogen). Expression data (presence–absence) in individual tissue samples were transformed into binary data, and non-parametric Spearman rank correlation tests were carried out for all possible pairwise comparisons of NITR genes.
Results
Sea bass NITRs identification and mapping
Shotgun sequencing of the two BAC clones yielded 2,021 reads for bassbac-18k1 (min Q20 length 300 bp, average length 649 bp) and 1,979 reads for bassbac-79e10 (min Q20 length 300 bp, average length 675 bp). Assembly of the data resulted in two contigs (136,804 and 10,268 bp with an average sequencing coverage of 8.9-fold) for bassbac-18k1 that could be ordered in a scaffold according to the BAC-ends sequences. For bassbac-79e10, the assembly yielded four contigs (3,184, 29,024, 100,505 and 4,881 bp with an average sequencing coverage of 9.7-fold) that were ordered into a scaffold by paired end information of plasmid reads and BAC end sequences. Both BACs share an overlapping region of ?13.5 kb and were manually aligned to build a single scaffold of 271,448 bp (including gaps). Average G+C content of the sequenced region was 40.5%.
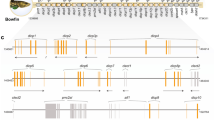
In silico analysis of the BAC-contig sequence revealed a NITR gene cluster in the European sea bass. Blast search using publicly available NITR protein sequences discovered 27 putative NITR genes (Dl1–Dl27). Three additional genes (Dl28–Dl30) were subsequently found based on their similarity to the first identified sea bass NITRs (Fig. 1). By using this approach, only portions of V, I (V/C2), TM and Cyt2 domains were identified. In order to experimentally validate in silico predictions, the full length of five transcripts (Dl8, Dl10, Dl20, Dl24, Dl25) and the whole coding regions of all the remaining sea bass NITR, cDNAs were amplified and sequenced. Three putative NITR genes were composed of only sparse domain fragments and never gave an amplification product from cDNA. These genes (Dl7, Dl18 and Dl27) were therefore considered as pseudogenes. Sea bass BAC contig and NITR sequences are available in GenBank under the accession numbers GQ385066 and GQ385039–GQ385065, respectively.

Genomic organisation of sea bass NITR gene cluster. Gene names are indicated; arrowhead indicates transcriptional orientation. Rectangles correspond to exons (L leader, V V domain, I I domain, TM transmembrane domain, Cyt1 and Cyt2 cytoplasmic domain, CP putative connecting peptide). In grey are indicated putative pseudogenes
Sea bass NITRs share conserved features
As reported for other teleosts, sea bass NITRs are related to Ig and TCR. Conserved amino acid positions, common to Ig and TCR genes, are found in either NITR V and/or I (V/C2) domains (Fig. 2). Eight conserved cysteine residues (Piyaviriyakul et al. 2007) are found in all sea bass NITRs. All NITRs identified so far in D. labrax can be classified as inhibitory based on the presence of one or two cytoplasmic ITIM or ITIM-like sequences as well as the absence of a charged transmembrane residue, with the exception of Dl15 where the cytoplasmic domains are missing altogether. No sea bass NITR displays activating cassettes such as ITSM (Yoder 2009) or ITAM-related sequences.

Sequence identity patterns in sea bass NITR V and I domains. ClustalW alignment of predicted translation product of sea bass V and I domains. Identical residues are highlighted in black; similar residues are highlighted in grey. The location of sequence regions corresponding to CDR1, CDR2 and CDR3 as well as J-related domains are indicated. Highly conserved Ig domain residues are designated in one-letter code; numbers correspond to International Immunogenetics Database (IMGT) designation (Lefranc et al. 2003). Conserved cysteines are indicated (asterisk) below the alignment
In accordance with the overall conservation of the main structural motifs, strong similarity of sea bass NITRs with homologues from other teleosts was confirmed also based on secondary structure analysis (Fig. 3). Using a comparative modelling approach, the highest similarity was found with the recently solved I. punctatus NITR10 and NITR11 (Cannon et al. 2008), which in turn demonstrated to be structurally similar to TCR in chain topology and dimerisation mode.

Homology model of sea bass NITR V domain. Three-dimensional models of sea bass NITR28 and NITR29 homodimers. Secondary structure of one subunit is depicted in gold, the other one in grey. CDR1 is highlighted in red, CDR2 is highlighted in green and CDR3 in blue. Black arrows indicate the four amino acid insertion in sea bass NITR 28. Putative connecting peptide is also shown
The NITR gene cluster is composed of multiple families
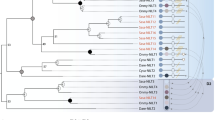
The 27 NITR genes identified are subdivided into 18 different V families, following a definition where a gene family comprises all sequences that share >70% predicted V domain amino acid identity (Electronic supplementary material (ESM), Supplementary file 1). These families differ in the number of ectodomains, genomic organisation, presence and location of J, J-like, ITIM and ITIM-like sequences. Phylogenetic analysis divided sea bass NITRs into three major clusters with high bootstrap confidence (Fig. 4). Group I (Fig. S1 in ESM, Supplementary file 2) is composed of 15 genes where V and I domains are encoded in a single exon. A similar genomic structure was already reported for all pufferfish NITR genes (Strong et al. 1999) and for some medaka ones (Desai et al. 2008). The only exceptions to this common organisation are represented by Dl6 where the I domain is missing and Dl8 which lacks the Cyt1 domain. In group II, the V and I domains are encoded by two separate exons. In both groups I and II, J regions with glycine bulges ([GXG] and [FGXG]) are present but located in the opposite order (Fig. S2 in ESM, Supplementary file 2). Group III genes (Fig. S3 in ESM, Supplementary file 2) are characterised by the presence of only one Ig-like (V-type) extracellular domain and the lack of J or J-like motifs. Within group III, four genes display an additional exon, between the V domain and the transmembrane region, which is 30 bp long in Dl15 and 42 bp in Dl28–30. This novel exon cannot be aligned between Dl15 and Dl28–30 NITRs, neither at the DNA nor at the protein level; thus, it seems to have been independently acquired in Dl15 and Dl28–30. Molecular modelling of Dl28 indicates that the novel exon encodes a putative connecting (stalk) peptide between the single Ig domain and the transmembrane region (Fig. 3).

Phylogenetic relationships between sea bass NITR V domains. Maximum likelihood tree of V domains encoded by 27 sea bass NITRs. Gene names are indicated. Bootstrap values are assigned to each interior branch; bootstrap values less than 50 are not shown. Branch lengths correspond to the number of aminoacid substitutions, with the scale indicated at the bottom of the figure. Generalised genomic organisations of each NITRs group is shown (not to scale). Rectangles correspond to exons (L leader, V V domain, I I domain, CP putative connecting peptide, TM transmembrane domain, Cyt1 and Cyt2 cytoplasmic domains)
Phylogenetic analyses suggest NITRs conform to the “birth-and-death” model of evolution
To set sea bass NITRs in a broader evolutionary perspective, a phylogenetic tree was constructed based on all publicly available NITR sequences (Fig. 5). It should be noted here that for some species included in the analysis (rainbow trout, channel catfish, Japanese flounder and green-spotted pufferfish), NITR genes have not yet been completely characterised. Nodes with bootstrap values >50% identified several gene clusters, although in most cases with moderate bootstrap support (<90%). The three most basal groups (nodes A, B, C), which contain nearly exclusively zebrafish, channel catfish, and trout NITRs, do not include any sea bass genes. All NITRs identified for this species are distributed in different clusters diverging at node D. The first group to differentiate, with good bootstrap support (node E, 85%), contains only NITRs showing a single Ig-like (V) domain, with a subgroup (node F, 99%) unique for acanthopterygian species (medaka, pufferfish, sea bass). However, not all fish “V-only” NITRs are included in cluster E. Channel catfish NITR5-11 (Ip5-11), all lacking the I domain, are clearly clustered in a different group (diverging at node A). Node G identifies the largest set of genes, comprising one cluster (node H) with exclusively genes in which the V and I domains are encoded by two separate exons and a second one (node J) that contains mainly NITRs with the V and I domains encoded in a single exon, without any intervening sequence. The majority of genes showing this genomic organisation appear to have a common origin (node N), although there are two notable exceptions. Medaka NITR24 and NITR10 (Ol24 and Ol10), as well as D. labrax NITR1 (Dl1), Spheroides nephelus NITR10 (Sn10) and one Tetraodon nigroviridis NITR (Tn72801), included in cluster L, are not comprised in the same group N. Alternative phylogenetic hypotheses, for instance forcing Dl1, Sn10 and Tn72801 within group N, were tested and statistically rejected (data not shown). Phylogenetic evidence therefore suggests that the intron spacing the two Ig-encoding exons might have been lost three times independently. NITR genes showing this genomic organisation have been found so far only in acanthopterigian species where they represent the majority of all identified NITRs (23/23 in pufferfish, 22/44 in medaka, 13/27 in sea bass). A detailed phylogenetic analysis based on nucleotide alignment of V- and I-encoding regions reveals that with the exception of Ol24, Ol10, Dl1, Sn10 and Tn72801, all these genes evolved through lineage-specific, independent expansions (ESM, Supplementary file 3).

Phylogenetic relationships between published NITR V domains. Published NITR sequences described from Southern pufferfish (S. nephelus, Sn), zebrafish (D. rerio, Dr), rainbow trout (Oncorhynchus mykiss, Om), channel catfish (I. punctatus, Ip), Japanese flounder (Paralichthys olivaceus, Po), medaka (O. latipes, Ol), T. nigroviridis (Tn) and European sea bass (D. labrax, Dl) were included in the alignment. Sequences are defined by the two-letter species abbreviation followed by the NITR gene number in that species. Only one NITR gene family member is incorporated; the other family members are indicated in parenthesis. NITR genes which encode a V and I domain in a single exon, V and I domain in two separate exons or only the V domain are highlighted with different colours. Sea bass NITR gene groups are also indicated. Bootstrap values are assigned to each interior branch; bootstrap values less than 50 are not shown. Branch lengths correspond to the number of amino acid substitutions, with the scale indicated at the bottom of the figure
NITRs experienced positive selection
A panel of different statistical tests was applied to four of these gene expansions, namely, the sea bass cluster of V-only NITRs (Dl10–15, Dl28–30) (group III in Fig. 4) and the three species-specific clusters of NITR genes with V and I domains encoded in a single exon (ESM, Supplementary file 3), in order to test whether gene duplications/expansions are associated with positive (divergent) selection. LRTs provide clear evidence that all four groups of NITRs experienced positive selection (ESM, Supplementary file 4), with high statistical confidence. For each dataset, four different approaches were then applied to identify codon sites that are most likely subject to positive selection (Table 1 and ESM, Supplementary files 5 and 6). When applying multiple methods, the use of high (e.g. 0.25) nominal ? levels followed by a consensus approach has been suggested (Kosakovsky Pond and Frost 2005). Here, although enforcing a more conservative ? level (0.05) and considering only sites that are predicted under positive selection by at least two or three methods (50% or 75% majority consensus (MC) rule), different amino acid positions were inferred to show adaptive changes. Nonetheless, and despite the small number of analysed sequences, for V-only NITR genes, eight positions under selection are identified with at least three methods and one with all four approaches (Table 1). Remarkably, seven of these sites are located in one putative complementarity determining region (CDR1, CDR2, CDR3; Fig. 2), and three are highly significant also according to the method that takes into account the different weight of amino acid substitutions. The eighth site, which is supported by all analyses, is immediately adjacent to CDR2. In Dl28, a four amino acid insertion separates this site under selection from CDR2, causing a substantial modification in the predicted protein structure (Fig. 3) and further diversifying V-only NITRs in the sea bass.
Proportionally, a smaller number of amino acid positions are inferred to have experienced adaptive selection in the remaining three gene expansions. Following either a 50% or a 75% MC rule, 29 and 4 positively selected positions are found in the sea bass (ESM, Supplementary file 5), 22 and 3 in the Southern pufferfish and six and two in medaka (ESM, Supplementary file 6), respectively. The most supported sites are observed in CDRs as well as other protein domains (cyt2, I).
Sea bass NITRs share conserved synteny
A comparative genomic analysis was carried out to identify genomic regions showing significant matches with sea bass NITRs. Regions isolated both in zebrafish and medaka correspond to those already described by Yoder and et al. (2002, 2004) and by Desai et al. (2008). In addition, our analysis identified the putative sea bass orthologues of FXR2, RBM4 and TRAF2 binding protein, three genes located in close proximity to NITR gene clusters in medaka chr. 18 and zebrafish chr. 7 (Fig. 6). A NITR gene cluster, situated in the stickleback chromosome/group IV has been already reported (Desai et al. 2008). In the present study, an additional NITR gene complex was found spanning three contigs (contig_7245-7247, ENSEMBL release 54) on G. aculeatus group VII. This cluster encompasses a region of ?250 kb and comprises at least 24 NITRs showing V and I domains encoded either by one or two exons (data not shown). Genes encoding RBM4, FXR2 and TRAF2-binding protein homologous were also found at the 3? of this region, indicating that the sea bass NITR cluster and the stickleback cluster located in group VII share conserved synteny along with those already reported on medaka chr. 18 and zebrafish chr. 7 (Fig. 6).

The synteny of sea bass NITR cluster is conserved in zebrafish, medaka and stickleback. Comparison of the European sea bass NITR gene cluster with the syntenic regions of zebrafish, medaka and stickleback. Genes (boxes) and the distances between them are not to scale
Expression analysis of sea bass NITRs
A preliminary survey on sea bass NITR gene expression was carried out using end-point RT-PCR on tissue samples taken from four different fish. Little or no expression was observed for Dl2, Dl4 and Dl16, whilst variation across individuals and tissues were noticed for the other NITR genes. Not surprisingly, NITRs are primarily detected in tissues that play a relevant role in immune responses. All NITRs are expressed in the head kidney and, with a few exceptions, in gills, gut and spleen. Weak (and variable across individuals) expression was observed in whole blood, liver and skeletal muscle (Table S1 in ESM, Supplementary file 7).
Non-parametric correlation tests on presence–absence data (Table S2 in ESM, Supplementary file 7) shows that for several gene pairs, mRNA expression is positively correlated, suggesting that in most cases, transcriptional regulation is partially conserved after duplication. However, correlation of expression patterns does not reflect well the phylogenetic/evolutionary relatedness of different NITR genes. The group of NITR genes Dl10–14 provides an interesting example on this issue. Based on the sea bass NITR phylogenetic tree (Fig. 4) and their genomic organisation (Fig. 1), the genes appear to have evolved from a single ancestral copy first through a gene duplication, giving rise to the ancestor of Dl10/Dl11and Dl12/13/14, respectively. A second round of duplication gave rise to Dl10 and Dl11, as well as to Dl14 and Dl12/Dl13. A final duplication event resulted in the emergence of Dl12 and Dl13. Comparison of NITR10 and NITR11 expression reveals 84% identity (27/32) across tissue/sample combinations, with a Spearman rho equal to 0.74 (p?<?0.00001), in agreement with the small phylogenetic distance between these two genes. On the other hand, between NITR12 and NITR13, only 56% identity (Spearman rho 0.24, p?=?0.17) is observed despite the two sequences being closely related. In this case, NITR13 gene expression seems to be under different selective constraints because the comparison between NITR12 and NITR14 shows a better overlap (68%) and a higher, marginally significant correlation (Spearman rho?=?0.33, p?=?0.06).
Discussion
NITR genes have been proposed to share functional features with mammalian natural killer receptors (NKRs), which can function to either inhibit or activate NK cell cytotoxicity and/or cytokine release (Yoder 2009). In common with other immune genes, both NKRs and NITRs appear to follow a “birth-and-death” model of evolution (Nei and Rooney 2005). This model assumes that, when two species are compared for a specific multi-gene family, most genes originate from a set of paralogous sequences present in the common ancestor through tandem duplications (“birth” process) after the divergence of the two lineages. On the other hand, several copies are lost through pseudogenization–deletion (“death” process; ESM, Supplementary file 8). In contrast to what happens in concerted evolution, a variable degree of sequence divergence is expected among family members as well as a certain proportion of pseudogenes depending on the selective forces acting on the specific gene family (Nei and Rooney 2005). In agreement with model predictions, three pseudogenes (10% of all isolated genes) are found in the sea bass, similar to what has been observed in other fish species, and three main gene groups are found, each one with a distinctive domain composition and genomic organisation (Fig. 4). Sequence identity >70% was considered a suitable criterion to define a gene family and allowed sea bass NITRs to be further divided into a large number of families (18), as already described for zebrafish, catfish and medaka NITRs (Yoder et al. 2001, 2004; Hawke et al. 2001; Desai et al. 2008). Diversification of sea bass NITR genes appear to have occurred through repeated tandem duplications, since most genes are organised in smaller, co-linear genomic subclusters which largely reflect their phylogenetic position (Figs. 1 and 4). The observed association between phylogenetic and physical clustering of members of different families suggests unequal crossing over as the major mechanism for their amplification. For instance, the sea bass Dl8 family includes three different genes (Dl8, Dl9 and Dl16) which are likely derived from a recent duplication event. Their encoded proteins are >85% identical, in particular Dl9 and Dl16 encode proteins with 95% identity, with intronic sequences showing >90% identity. Similar evidence comes from the Dl28 family (Dl28–30) where exon sequences are 85–91% identical, whilst all introns but one could be aligned.
When set in a larger evolutionary perspective, the different groups (I–III) of sea bass NITRs again appear to conform to the “birth-and-death” model predictions. The origin of group I can be traced back to a minimal set of two genes in the common ancestor of acanthopterygian fish (medaka, pufferfish, sea bass). One of these two genes has been retained in single copy in T. nigroviridis, S. nephelus and D. labrax (grouped under node L in Fig. 5), whereas it has been lost in O. latipes. The other ancestral gene underwent lineage-specific, independent expansions (node N in Fig. 5 and, in details, ESM, Supplementary file 3). Group II is the least numerous in the sea bass, and it appears that homologous genes in other teleosts (node H in Fig. 5) have experienced a limited number of duplication events as well. Group III is characterised by lack of the I domain-encoding exon, which is replaced by a novel exon in four genes (Dl15, Dl28–30). Whilst V-only NITRs have been reported in several other species (channel catfish, zebrafish, medaka), the acquisition of an additional exon has been observed so far only in I. punctatus NITR5-11. As discussed earlier, this acquisition is likely to have occurred independently three times. Two mechanisms have been proposed for the evolution of novel exons. The first one, “exonization” of an intronic region, requires the evolution of a splice site surrounding the recruited genomic sequence (Sorek 2007). The second one, “exon shuffling”, promotes the gain of an exon through crossing over between non-homologous genes, causing the insertion of an exon from a “donor” gene into a preexisting intron of a recipient gene (Patthy 2008). Under the exonization model, a sequence with some similarity with the novel exon should be retained in the corresponding introns of related genes that did not acquire an additional exonic sequence. However, genomic alignment of intron 2 of Dl10–14 shows no such evidence. Exon shuffling is therefore the most likely mechanism for novel exon acquisition, as already reported for KIR genes (Rajalingam et al. 2004).
The function of the peptide encoded in the extra exon is not clear at present. No functional domain or motif has been identified. Homology modelling (Fig. 3) maps it to the “stalk” region reported for the channel catfish NITR11 (Hawke et al. 2001; Cannon et al. 2008), and further analysis using TMHMM v2.0 confirms its extracellular localisation, suggesting it might encode a connecting peptide. Connecting peptides are commonly found in other immunoglobulin-like molecules such as NILTs in teleosts (Stet et al. 2005; Østergaard et al. 2008) as well as mammalian KIRs and ILTs (Torkar et al. 1998; Martin et al. 2000). In NILT genes, it has been suggested that this region delimits a potentially accessible pocket playing a crucial role for ligand binding. Whether the repeated acquisition of the stalk-encoding exon has any functional meaning in fish NITRs remains to be further elucidated.
Convergent evolution towards similar genomic organisations (number of exons, intron–exon structure) seems to be a more general feature of fish NITR genes. At present, it is not clear which was the original organisation in the ancestral NITR. However, if it is postulated that a structure with six exons and two Ig domains was the ancestral state for the genomic structure of NITR genes, then loss of I, TM and cytoplasmic domains, as well as of the intron spacing V-encoding and I-encoding exons appears to have occurred several times independently. Modular organisation and evolution through domain shuffling provide additional evidence for the similarity between NITRs and NKRs, particularly KIRs where two to three Ig (C) domains are alternatively present and long or short forms, either including or excluding a long cytoplasmic tail, are found. Both activating and inhibitory alleles of KIR 3DL1/S1 locus have been found to be under positive, directional selection in some human populations based on a codon model ML analysis (Norman et al. 2007). Selected amino acid positions were located in the two Ig domains (D1 and D2) that bind MHC-I proteins as well as on the third Ig domain (D0), which is supposed to enhance binding capacity. In the present study, using a similar approach, four clusters of NITR genes, in three different species, were demonstrated to have experienced positive, divergent selection. This is the first evidence that the extraordinary diversity of NITR genes reported in several fish lineages might be, at least in part, the consequence of adaptive selection favouring the evolution of a highly diverse array of molecules. Maximum likelihood methods for detecting positive selection have some limitations (Nozawa et al. 2009), yet they represent the most rigorous approach available to date to assess which evolutionary forces (negative–positive–(nearly) neutral) have been acting on a group of sequences (Anisimova and Kosiol 2009). Lineage-specific gene expansions have already been suggested to be a peculiar, recurrent feature of fish NITRs (Yoder 2009). Evidence presented here further supports the important role of this phenomenon, indicating that repeated gene duplications within NITR families likely provide the raw material for positive, diversifying selection to work on. This observation is in perfect agreement with genome-wide surveys conducted to assess the role of positive selection in gene evolution, which have already pointed out that positively selected genes are significantly enriched for roles in defence/immunity (e.g. Bustamante et al. 2005; Kosiol et al. 2008). Whilst these studies have generally excluded recent gene families, Han et al. (2009) have reported that recently duplicated genes show more frequently evidence of positive selection compared to single-copy genes.
The application of probabilistic codon models can also predict which sites are most likely under positive selection in a specific protein (Anisimova and Kosiol 2009). Although not conclusive and requiring further experimental studies, the consensus among different methods suggests that positively selected codons are often located in putative ligand-binding regions. Whilst ligands for NITRs remain to be identified, a recent study (Cannon et al. 2008) clearly demonstrated that a single amino acid substitution in CDR1 of channel catfish NITRs is sufficient to acquire/abrogate recognition of an allogeneic B cell target. Also, the close similarity in protein structure between NITR and TCR/Ig-LC suggests that CDR1-3 might have a similar role of ligand recognition in NITRs. Thus, it is not unexpected to find positively selected sites in these regions which might be under diversifying selection as already shown for recombining receptors of adaptive immunity. Evidence of positive Darwinian selection in the extracellular V domain of NITR genes also fits well with the hypothesis that fish NITRs are functionally homologous to mammalian KIRs, which in primates mediate self-recognition of NK cells under the missing self model. Inhibitory, self-recognising KIRs are supposed to be under diversifying selection to keep pace with the enormous diversity of their ligands, encoded by hundreds of different alleles at the MHC-I loci. Although evolutionary strategies are different across teleosts, ranging from a single classical MHC-I locus with highly polymorphic alleles in the rainbow trout (Phillips et al. 2003) to tens–hundreds of loci in the Nile tilapia (Sato et al. 2006) and the European sea bass (L. Bargelloni, unpublished observations), fish show high intraspecific diversity of MHC-I proteins as observed in mammals. Finally, it should be noticed that codon models likely underestimate coding sequence variation between NITR sequences since insertions and/or deletions cannot be incorporated in such models. For instance, sea bass NITR28 shows a four amino acid insertion at the end of CDR2 where the most significant positively selected site was also found (Table 1), with a visible effect on predicted protein structure (Fig. 3).
In addition to the putative ligand recognition region, there is evidence that a few sites in the cytoplasmic portion of the protein might be positively selected. The regulatory role of the cytoplasmic tail appears to be conserved compared to NKRs (Yoder et al. 2001, 2007; Wei et al. 2007). Activating or inhibitory signals are transduced through binding of intracellular partner proteins (e.g. DAP12, SHPs). Thus, it might be possible that amino acid changes in the cytoplasmic tail modify the specificity and/or affinity of these protein–protein interactions, modulating signal transduction between different NITRs.
Comparative genomics of fish NITRs yields additional evidence in favour of a “birth-and-death” model of evolution. Two major NITR clusters were found in zebrafish on chromosomes 7 and 14 (Yoder et al. 2001, 2008), medaka chromosomes 18 and 10 (Desai et al. 2008) and stickleback groups VII and IV (Desai et al. 2008; this study), which are onhologs (chromosome pairs deriving from a single ancestral chromosome after a whole genome duplication; Nakatani et al. 2007; Negrisolo et al., submitted). As already pointed out (Yoder 2009), this evidence suggests the presence of a substantial set of NITR genes in the common ancestor of all teleost fish before the occurrence of the teleost-specific whole genome duplication event (Nakatani et al. 2007). Once duplicated, the original cluster was retained in two copies, although individual NITR genes have been alternatively lost, retained or expanded mostly independently in each fish lineage examined so far. For instance, a closer examination of NITRs grouped under node H (Fig. 5) reveals that sea bass group II are more closely related to zebrafish NITR 6a,b and 8 (chr. 7) than with medaka Ol 6a,b,c and Ol18-22 (chr. 22) despite the fact that medaka and seabass are phylogenetically closer than sea bass with zebrafish. These zebrafish and sea bass genes, however, are located on orthologous clusters, whereas the medaka ones are positioned on the corresponding onholog. This indicates that orthologs of sea bass group II genes have been retained in zebrafish, whilst only their paralogs are found in medaka. All these evidence, together with the shared synteny found between sea bass NITR cluster and stickleback/medaka/zebrafish chromosomes IV/10/14 (Fig. 6), suggest that in sea bass as well might exist at least one more NITR cluster not yet identified.
In agreement with observations in other species (Piyaviriyakul et al. 2007; Yoder et al. 2002), NITR genes are predominantly expressed in tissues playing a relevant role in the immunological response, whilst gene-specific variation was noticed in other tissues. Weak or null expression observed on blood samples might be partially explained by the fact that whole blood was used rather than pheripheral blood leucocytes due to the limited amount of available biological material.
The results of expression analysis presented here are preliminary and await further quantitative studies with qRT-PCR, possibly on cell line-specific RNAs rather than whole tissue samples. Nevertheless, moderate correlation between gene pairs in presence–absence expression data suggests that in most cases, transcriptional regulation is partially conserved after duplication. However, expression pattern does not reflect well the phylogenetic/evolutionary relatedness of different NITR genes, thereby revealing differential selective constraints on gene expression and providing further room for evolutionary diversification of NITR genes.
In summary, 27 NITR genes and three pseudogenes were identified and analysed in the European sea bass, providing significant insights into the evolution and function of NITR genes in bony fish. Based on the results reported here, in addition to those collected so far in other species, the following conclusions can be drawn: (1) starting from two gene clusters in the common ancestor of all teleosts, NITRs have evolved following a “birth-and-death” model of evolution, with lineage-specific gain and loss of individual family members, (2) recurrent events of domain shuffling and exon/intron acquisition/deletion contributed to the remarkable diversity of domain and genomic organisation of fish NITRs, (3) repeated duplications have generated largely species-specific gene expansions, which have been demonstrated to be under positive Darwinian selection in at least four independent cases, (5) directional selection seems to significantly affect putative ligand binding regions, confirming the predicted function of NITR proteins and (5) differential expression might be a further dimension of NITR diversity.
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21(9):2104–2105. doi:10.1093/bioinformatics/bti263
Anisimova M, Kosiol C (2009) Investigating protein-coding sequence evolution with probabilistic codon substitution models. Mol Biol Evol 26(2):255–271. doi:10.1093/molbev/msn232
Bléry M, Olcese L, Vivier E (2000) Early signaling via inhibitory and activating NK receptors. Hum Immunol 61(1):51–64. doi:10.1016/S0198-8859(99)00157-3
Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT et al (2005) Natural selection on protein-coding genes in the human genome. Nature 437:1153–1157. doi:10.1038/nature04240
Campanella JJ, Bitincka L, Smalley J (2003) MatGAT: an application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinformatics 4:29
Canavez F, Young NT, Guethlein LA, Rajalingam R, Khakoo SI, Shum BP, Parham P (2001) Comparison of chimpanzee and human leukocyte Ig-like receptor genes reveals framework and rapidly evolving genes. J Immunol 167:5786–5794
Cannon JP, Haire RN, Magis AT, Eason DD, Winfrey KN, Hernandez Prada JA, Bailey KM, Jakoncic J, Litman GW, Ostrov DA (2008) A bony fish immunological receptor of the NITR multigene family mediates allogeneic recognition. Immunity 29:228–237. doi:10.1016/j.immuni.2008.05.018
Chou HH, Holmes MH (2001) DNA sequence quality trimming and vector removal. Bioinformatics 17:1093–1104. doi:10.1093/bioinformatics/17.12.1093
Desai S, Heffelfinger AK, Orcutt TM, Litman GW, Yoder JA (2008) The medaka novel immune-type receptor (NITR) gene clusters reveal an extraordinary degree of divergence in variable domains. BMC Evol Biol 8:177. doi:10.1186/1471-2148-8-177
Doron-Faigenboim A, Pupko T (2006) A combined empirical and mechanistic codon model. Mol Biol Evol 24:388–397. doi:10.1093/molbev/msl175
Evenhuis J, Bengtén E, Snell C, Quiniou SM, Miller NW, Wilson M (2007) Characterization of additional novel immune type receptors in channel catfish, Ictalurus punctatus. Immunogenetics 59:661–671. doi:10.1007/s00251-007-0230-x
Ewing B, Green P (1998) Base-calling of automated sequencer traces using PHRED. II. Error probabilities. Genome Res 8:186–194
Felsenstein J (2004) Inferring phylogenies. Sinauer, Sunderland
Guindon S, Gascuel O (2003) A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704. doi:10.1080/10635150390235520
Han MV, Demuth JP, McGrath CL, Casola C, Hahn MW (2009) Adaptive evolution of young gene duplicates in mammals. Genome Res 19(5):859–867. doi:10.1101/gr.085951.108
Hawke NA, Yoder JA, Haire RN, Mueller MG, Litman RT, Miracle AL, Stuge T, Shen L, Miller N, Litman GW (2001) Extraordinary variation in a diversified family of immune-type receptor genes. Proc Natl Acad Sci USA 98:13832–13837. doi:10.1073/pnas.231418598
Jobb G, von Haeseler A, Strimmer K (2004) TREEFINDER: a powerful graphical analysis environment for molecular phylogenetics. BMC Evol Biol 4:18
Kock H, Fischer U (2008) A novel immunoglobulin-like transcript from rainbow trout with two Ig-like domains and two isoforms. Mol Immunol 45(6):1612–1622. doi:10.1016/j.molimm.2007.10.005
Kosakovsky Pond SL, Frost SD (2005) Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22(5):1208–1222. doi:10.1093/molbev/msi105
Kosiol C, Vinar T, da Fonseca RR, Hubisz MJ, Bustamante CD, Nielsen R, Siepel A (2008) Patterns of positive selection in six mammalian genomes. PLoS Genet 4(8):e1000144. doi:10.1371/journal.pgen.1000144
Lefranc MP, Pommié C, Ruiz M, Giudicelli V, Foulquier E, Troung L, Thouvenin-Contet V, Lefranc G (2003) IMGT unique numbering for immunoglobulin an T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol 27(1):55–77. doi:10.1016/S0145-305X(02)00039-3
Lewis SE, Searle SMJ, Harris N, Gibson M, Iyer V, Richter J, Wiel C, Bayraktaroglu L, Birney E, Crosby MA, Kaminker JS, Matthews BB, Prochnik SE, Smith CD, Tupy JL, Rubin GM, Misra S, Mungall CJ, Clamp ME (2002) Apollo: a sequence annotation editor. Genome Biol 3(12):research0082
Litman GW, Hawke NA, Yoder JA (2001) Novel immune-type receptor genes. Immunol Rev 181:250–259
Martin AM, Freitas EM, Witt CS, Christiansen FT (2000) The genomic organization and evolution of the natural killer immunoglobulin-like receptor (KIR) gene cluster. Immunogenetics 51:268–280. doi:10.1007/s002510050620
Martinez-Borra J, Khakoo SI (2008) Speed and selection in the evolution of killer-cell immunoglobulin-like receptors. Int J Immunogenet 35:89–96. doi:10.1111/j.1744-313X.2008.00756.x
Meyaard L, Adema GJ, Chang C, Woollatt E, Sutherland GR, Lanier LL, Phillips JH (1997) LAIR-1, a novel inhibitory receptor expressed on human mononuclear leukocytes. Immunity 7(2):283–290. doi:10.1016/S1074-7613(00)80530-0
Nakatani Y, Takeda H, Kohara Y, Morishita S (2007) Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res 17(9):1254–1265. doi:10.1101/gr.6316407
Nei M, Rooney AP (2005) Concerted and birth-and-death evolution of multigene families. Annu Rev Genet 39:121–152. doi:10.1146/annurev.genet.39.073003.112240
Nelson JS (2006) Fishes of the world, 4th edn. Wiley, Hoboken
Nielsen R, Yang ZH (1998) Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936
Norman PJ, Abi-Rached L, Gendzekhadze K, Korbel D, Gleimer M, Rowley D, Bruno D, Carrington CV, Chandanayingyong D, Chang YH, Crespí C, Saruhan-Direskeneli G, Fraser PA, Hameed K, Kamkamidze G, Koram KA, Layrisse Z, Matamoros N, Milà J, Park MH, Pitchappan RM, Ramdath DD, Shiau MY, Stephens HA, Struik S, Verity DH, Vaughan RW, Tyan D, Davis RW, Riley EM, Ronaghi M, Parham P (2007) Unusual selection on the KIR3DL1/S1 natural killer cell receptor in Africans. Nat Genet 39(9):1092–1099. doi:10.1038/ng2111
Nozawa M, Suzuki Y, Nei M (2009) Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc Natl Acad Sci USA 106(16):6700–6705. doi:10.1073/pnas.0901855106
Østergaard AE, Martin SAM, Wang T, Stet RJM, Secombes CJ (2008) Rainbow trout (Oncorhynchus mykiss) possess multiple novel immunoglobulin-like transcripts containing either an ITAM or ITIMs. Dev Comp Immunol 33(4):525–532. doi:10.1016/j.dci.2008.10.002
Patthy L (2008) Exons: shuffling. In Encyclopedia of life sciences (ELS). Wiley, Chichester. doi:10.1038/npg.els.0005087
Phillips RB, Zimmerman A, Noakes MA, Palti Y, Morasch MRW, Eiben L, Ristow SS, Thorgaard GH, Hansen JD (2003) Physical and genetic mapping of the rainbow trout major histocompatibility regions: evidence for duplication of the class I region. Immunogenetics 55:561–569. doi:10.1007/s00251-003-0615-4
Piyaviriyakul P, Kondo H, Hirono I, Aoki T (2007) A novel immune-type receptor of Japanese flounder (Paralichthys olivaceus) is expressed in both T and B lymphocytes. Fish Shellfish Immunol 22:467–476. doi:10.1016/j.fsi.2006.05.007
Pond SL, Frost SD (2005) Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21(10):2531–2533. doi:10.1093/bioinformatics/bti320
Randelli E, Buonocore F, Casani D, Fausto AM, Scapigliati G (2009) An “immunome” gene panel for transcriptomic analysis of immune defence activities in the teleost sea bass (Dicentrarchus labrax L.): a review. Ital J Zool 76(2):146–157. doi:10.1080/11250000802572531
Rajalingam R, Parham P, Abi-Rached L (2004) Domain shuffling has been the main mechanism forming new hominoid killer cell Ig-like receptors. J Immunol 172(1):356–369
Sali A, Blundell TL (1993) Comparative protein modelling by satisfaction of spatial retraints. J Mol Biol 234:779–815. doi:10.1006/jmbi.1993.1626
Samadiris J, Colonna M (1997) Cloning of novel immunoglobulin superfamily receptors expressed on human myeloid and lymphoid cells: structural evidence for new stimulatory and inhibitory pathways. Eur J Immunol 27(3):660–665
Sato A, Dongak R, Hao L, Takezaki N, Shintani S, Aoki T, Klein J (2006) MHC class I genes of the cichlid fish Oreochromis niloticus. Immunogenetics 58:917–928. doi:10.1007/s00251-006-0151-0
Scapigliati G, Romano N, Buonocore F, Picchietti S, Baldassini MR, Prugnoli D, Galice A, Meloni S, Secombes CJ, Mazzini M, Abelli L (2002) The immune system of sea bass, Dicentrarchus labrax, reared in aquaculture. Dev Comp Immunol 26(2):151–160
Sorek R (2007) The birth of new exons: mechanisms and evolutionary consequences. RNA 13(10):1603–1608. doi:10.1016/S0145-305X(01)00057-X
Stern A, Doron-Faigenboim A, Erez E, Martz E, Bacharach E, Pupko T (2007) Selecton 2007: advanced models for detecting positive and purifying selection using a Bayesian inference approach. Nucleic Acids Res 35:W506–W511. doi:10.1093/nar/gkm382
Stet RJM, Hermsen T, Westphal AH, Jukes J, Engelsma M, Verburg-van Kemenade BML, Dortmans J, Aveiro J, Savelkoul HFJ (2005) Novel immunoglobulin-like transcripts in teleost fish encode polymorphic receptors with cytoplasmic ITAM or ITIM and a new structural Ig domain similar to the natural cytotoxicity receptor NKp44. Immunogenetics 57:77–89. doi:10.1007/s00251-005-0771-9
Strong SJ, Mueller MG, Litman RT, Hawke NA, Haire RN, Miracle AL, Rast JP, Amemiya CT, Litman GW (1999) A novel multigene family encodes diversified variable regions. Proc Natl Acad Sci USA 96:15080–15085. doi:10.1073/pnas.96.26.15080
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22(22):4673–4680. doi:10.1093/nar/22.22.4673
Torkar M, Norgate Z, Colonna M, Trowsdale J, Wilson MJ (1998) Isotypic variation of novel immunoglobulin-like transcript/killer cell inhibitory receptor loci in the leukocyte receptor complex. Eur J Immunol 28:3959–3967. doi:10.1002/(SICI)1521-4141(199812)28:12<3959::AID-IMMU3959>3.0.CO;2-2
Wei S, Zhou JM, Chen X, Shah RN, Liu J, Orcutt TM, Traver D, Djeu JY, Litman GW, Yoder JA (2007) The zebrafish activating immune receptor Nitr9 signals via Dap12. Immunogenetics 59(10):813–821. doi:10.1007/s00251-007-0250-6
Wolfsberg TG, Madden TL (2001) Sequence similarity searching using the BLAST family of programs. Curr Protoc Mol Biol. 19, Unit 19.3. doi:10.1002/0471142727.mb1903s46
Yang Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24(8):1586–1591. doi:10.1093/molbev/msm088
Yang Z, Wong WS, Nielsen R (2005) Bayes empirical bayes inference of amino acid sites under positive selection. Mol Biol Evol 22(4):1107–1118. doi:10.1093/molbev/msi097
Yang ZH, Nielsen R, Goldman N, Pedersen AMK (2000) Codon substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449
Yoder JA (2009) Form, function and phylogenetics of NITRs in bony fish. Dev Comp Immunol 33:135–144. doi:10.1016/j.dci.2008.09.004
Yoder JA, Mueller MG, Wei S, Corliss BC, Prather DM, Willis T, Litman RT, Djeu JY, Litman GW (2001) Immune type receptor genes in zebrafish share genetic and functional properties with genes encoded by the mammalian lymphocyte receptor cluster. Proc Natl Acad Sci USA 98:6771–6776
Yoder JA, Mueller MG, Nichols KM, Ristow SS, Thorgaard GH, Ota T, Litman GW (2002) Cloning novel immune-type inhibitory receptors from the rainbow trout, Oncorhynchus mykiss. Immunogenetics 54:662–670. doi:10.1007/s00251-002-0511-3
Yoder JA, Litman RT, Mueller MG, Desai S, Dobrinski KP, Montgomery JS, Buzzeo MP, Ota T, Amemiya CT, Trede NS, Wei S, Djeu JY, Humphray S, Jekosch K, Hernandez Prada JA, Ostrov DA, Litman GW (2004) Resolution of the novel immune-type receptor gene cluster in zebrafish. Proc Natl Acad Sci USA 101:15706–15711. doi:10.1073/pnas.0405242101
Yoder JA, Orcutt TM, Traver D, Litman GW (2007) Structural characteristics of zebrafish orthologs of adaptor molecules that associate with transmembrane immune receptors. Gene 401:154–164. doi:10.1016/j.gene.2007.07.014
Yoder JA, Cannon JP, Litman RT, Murphy C, Freeman JL, Litman GW (2008) Evidence for a transposition event in a second NITR gene cluster in zebrafish. Immunogenetics 60:257–265. doi:10.1007/s00251-008-0285-3
Yokoyama WM, Plougastel BFM (2003) Immune functions encoded by the natural killer gene complex. Nat Rev Immunol 3(4):304–316. doi:10.1038/nri1055
Acknowledgements
We thank Fabio Borghesan for providing sea bass individuals. We thank also Enrico Negrisolo for very helpful discussion and advice on phylogenetic analyses. This work was partially supported by EU project Aquagenome–genomics in fish and shellfish: From research to aquaculture.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary file 1
Comparison by pairwise alignments of the V domains from sea bass NITRs (% amino acid identity and similarity). (XLS 41 kb)
Supplementary file 2
Sea bass NITRs overall organisation. Peptide sequence alignments of NITR genes belonging to group I (Fig. S1), group II (Fig. S2) and group III (Fig. S3). (PDF 859 kb)
Supplementary file 3
Phylogenetic analysis of species-specific expansion across intronless NITRs of different species. Rooted phylogenetic tree showing the relationships between published NITR V and I domains. Published NITR nucleic sequences described from Southern pufferfish (Spheroides nephelus, Sn), medaka (Oryzias latipes, Ol), Tetraodon nigroviridis (Tn) and European sea bass (Dicentrarchus labrax, Dl) were included in the alignment. Sequences are defined by the two-letter species abbreviation followed by the NITR gene number in that species. Bootstrap values are assigned to each interior branch; bootstrap values less than 50 are not shown. Species-specific expansions are indicated. (PDF 232 kb)
Supplementary file 4
Tests for positive selection at NITR genes. (PDF 143 kb)
Supplementary file 5
Sea bass group I NITR amino acid sites under positive selection identified using different methods. (PDF 16 kb)
Supplementary file 6
Medaka and Southern pufferfish NITR amino acid sites under positive selection identified using different methods. (PDF 55 kb)
Supplementary file 7
NITR mRNA expression data (Table S1) and Spearman correlation of NITR expression patterns (Table S2). (PDF 54 kb)
Supplementary file 8
Example of “birth-and-death” model of evolution of a multigene family. ? indicates pseudogenes. (PDF 69 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Ferraresso, S., Kuhl, H., Milan, M. et al. Identification and characterisation of a novel immune-type receptor (NITR) gene cluster in the European sea bass, Dicentrarchus labrax, reveals recurrent gene expansion and diversification by positive selection. Immunogenetics 61, 773–788 (2009). https://doi.org/10.1007/s00251-009-0398-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-009-0398-3