
Abstract
Aims/hypothesis
Orexin A (OXA) modulates body weight, food intake and energy expenditure. In vitro, OXA increases PPARγ (also known as PPARG) expression and inhibits lipolysis, suggesting direct regulation of lipid metabolism. Here, we characterise the metabolic effects and mechanisms of OXA action in adipocytes.
Methods
Isolated rat adipocytes and differentiated murine 3T3-L1 adipocytes were exposed to OXA in the presence or absence of phosphoinositide 3-kinase (PI3K) inhibitors. Pparγ expression was silenced using small interfering RNA. Glucose uptake, GLUT4 translocation, phosphatidylinositol (3,4,5)-trisphosphate production, lipogenesis, lipolysis, and adiponectin secretion were measured. Adiponectin plasma levels were determined in rats treated with OXA for 4 weeks.
Results
OXA PI3K-dependently stimulated active glucose uptake by translocating the glucose transporter GLUT4 from cytoplasm into the plasma membrane. OXA increased cellular triacylglycerol content via PI3K. Cellular triacylglycerol accumulation resulted from increased lipogenesis as well as from a decrease of lipolysis. Adiponectin levels in chow- and high-fat diet-fed rats treated chronically with OXA were increased. OXA stimulated adiponectin expression and secretion in adipocytes. Both pharmacological blockade of peroxisome proliferator-activated receptor γ (PPARγ) activity or silencing Pparγ expression prevented OXA from stimulating triacylglycerol accumulation and adiponectin production.
Conclusions/interpretation
Our study demonstrates that OXA stimulates glucose uptake in adipocytes and that the evolved energy is stored as lipids. OXA increases lipogenesis, inhibits lipolysis and stimulates the secretion of adiponectin. These effects are conferred via PI3K and PPARγ2. Overall, OXA’s effects on lipids and adiponectin secretion resemble that of insulin sensitisers, suggesting a potential relevance of this peptide in metabolic disorders.
Similar content being viewed by others

Introduction
Orexin A (OXA) and orexin B are two isoforms derived from a common precursor peptide [1]. Both isoforms interact with two different G-protein-coupled receptor subtypes, orexin 1 (OXR1) and orexin 2 (OXR2) [1]. OXA binds with an approximately 10-fold higher affinity to OXR1 compared with OXB, whereas OXR2 binds both isoforms with similar affinity [1]. Orexin regulates food intake and body weight by acting in the central nervous system [1–3]. Intraventricular injection of orexin acutely leads to hyperphagia [1], whereas the deletion of orexin or the cognate receptor results in late-stage obesity, despite hypophagia [2]. In contrast, overproduction of orexin in mice protects from diet-induced obesity as a result of increased energy expenditure [4].
Low levels of orexin are found in most narcoleptic patients [5, 6]. In accordance with observations in animals, narcoleptic patients are obese and have increased incidence of type 2 diabetes mellitus [6, 7]. In addition, morbidly obese non-narcoleptic patients have lower plasma orexin levels than non-obese or moderately obese individuals [8], suggesting that OXA regulates body weight.
Outside the brain, orexin and its receptors have been detected in, for example, the adrenal glands, pituitary, endocrine islets and gut [9]. Orexin increases the secretion of cortisol, adrenaline (epinephrine) and growth hormone, and modulates glucose uptake from the gut. Orexin regulates the function of the endocrine pancreas, although discrepant findings have been reported. In some studies OXA inhibited insulin secretion and also stimulated glucagon secretion [10, 11]. We and others have demonstrated that OXA increases insulin secretion in vivo and in vitro, and inhibits the secretion of glucagon [12–14]. In mice with streptozotocin-induced diabetes, OXA lowered blood glucose levels [15].
A recent study in mice overexpressing orexin and orexin receptors demonstrated that orexin can ameliorate insulin resistance in a diet-induced obesity model [4], whereas orexin-deficient mice are insulin-resistant [16]. Through increased leptin sensitivity, OXA was able to improve insulin sensitivity in this study [4].
Obesity is a major risk factor for type 2 diabetes mellitus. Adipose tissue is a source of cytokines, which influence insulin sensitivity. The presence of orexin receptors was recently shown in 3T3-L1 cells (pre-adipocytes) and in adipocytes. OXA stimulated the proliferation of 3T3-L1 pre-adipocytes [17] and increased PPARγ2 expression in isolated human subcutaneous adipose tissue [18]. In omental adipocytes, OXA inhibited the expression of HSL (also known as LIPE) mRNA and the release of glycerol [18], indicating that OXA regulates lipid metabolism. Since OXA increases the expression of PPARγ2, a target receptor for treating insulin resistance in type 2 diabetes mellitus, it is possible that this mechanism may be responsible for the previously reported OXA-associated improvement of insulin sensitivity [4]. From previous studies using a heterologous expression system [19], it is known that OXA is able to enhance glucose uptake. Based on these findings, we investigate in the current study the role of OXA in regulating glucose uptake as well as the metabolic and endocrine consequence of the interaction of OXA with PPARγ-dependent processes, using isolated primary rat adipocytes and mature 3T3-L1 adipocytes.
Methods
Experimental animals
Adult male Wistar rats (200 ± 30 g, 10 weeks of age) were purchased from Lab Animal Breeding (Brwinow, Poland) and allowed to recover for 2 weeks.
In vivo study
Animals were fed a high-fat diet (HFD; 60% energy from fat; Diet No. 2127, Provimi Kliba AG Kliba Nafag, Kaiseraugst, Switzerland) or a standard diet for 5 weeks. Alzet osmotic pumps (model 2ML4; Durect, Cupertino, CA, USA) prefilled with OXA (0.134 mg per pump) were then placed s.c. into animals. OXA (50 pmol/h) or 0.9% NaCl (vehicle) was continuously delivered i.p. for 4 weeks. Plasma concentrations of OXA and body weights of rats treated with vehicle or OXA are shown in electronic supplementary material (ESM) Fig. 1a, b.
To insert the pumps, a small midline incision was made in the skin below the rib cage of the animal. Another small incision in the abdominal muscle was made directly under the cutaneous incision. The pump was inserted, flow moderator first, into the peritoneal cavity. The muscle and skin incisions were closed with sutures or wound clips.
The experimental protocols were approved by the Local Ethics Commission for Investigation on Animals, Poznań University of Life Sciences. The Principles of Laboratory Animal Care given in NIH publication No. 85-23, revised 1985 (http://grants1.nih.gov/grants/olaw/references/phspol.htm) were followed, as were specific national laws.
Reagents
Unless otherwise stated, all reagents were purchased from Sigma-Aldrich (Deisenhofen, Germany). Antibodies and their dilutions are listed in ESM Table 1. LY294002 was from Calbiochem (San Diego, CA). The non-thiazolidinedione peroxisome proliferator-activated receptor γ (PPARγ) activator (nTZD) indoleacetic acid (2-(2-(4-phenoxy-2-propylphenoxy)ethyl) indole-5-acetic acid) was kindly provided by D. von Langen and M. Kress of Merck Research Laboratories (Rahway, NJ, USA) [20]. 2-Deoxy-d-[2,6-3H]glucose and [U-14C]glucose were from Amersham Pharmacia Biotech (Little Chalfont, UK). Medium and supplements for cell culture were from Gibco Invitrogen (Karlsruhe, Germany). The PPARγ antagonist bisphenol A diglycidyl ether (BADGE) was from Fluka Chemicals (St Louis, MO, USA).
Isolation of rat adipocytes
Primary adipocytes were isolated from epididymal fat pads of male adult Wistar rats, as described by Rodbell [21]. Tissue pooled from two or three rats was purified from blood vessels, dried and minced with scissors. Fragmented tissue was digested in KRB HEPES buffer (KRBH) containing 3 mg/ml collagenase type II, 3% BSA and 5 mmol/l glucose for 45 min at 37°C in a shaking water-bath. Cells were then filtered through a nylon mesh (250 μm) and washed with KRBH. Adipocytes were counted using a Bürker–Türk counter chamber.
3T3-L1 cell culture and differentiation procedure
3T3-L1 fibroblasts purchased from ATCC (LGC Standards, Wesel, Germany) were maintained in a standard growth medium (DMEM, 10% [vol./vol.] FCS, 100 kU/l penicillin, 100 mg/l streptomycin) at 37°C in a humidified atmosphere (5% CO2, 95% O2). Differentiation into adipocytes was performed according to standard protocols [22]. All experiments were performed between the sixth and tenth day after induction of differentiation. Successful differentiation was evaluated by morphology and by measuring the mRNA expression of Fabp4 (>1,000-fold increase vs non-differentiated cells).
Real-time PCR
Total RNA was isolated using an RNeasy Mini Kit (Qiagen, Hilden, Germany). First, cDNA was generated from 2 μg of RNA using the ImProm-II Reverse Transcription System (Promega, Madison, WI, USA). Real-time PCR was performed with gene-specific intron spanning primers. Each real-time PCR reaction (in duplicate) containing LightCycler Fast Start DNA Master SYBR Green I or LightCycler TaqMan Master Kit and 5 μl cDNA diluted 1:10 in a total volume of 20 μl was carried out in a Roche LightCycler 2.0 (Roche Diagnostics, Penzberg, Germany). The specificity of PCR products was monitored on the basis of melting curve analysis. Relative quantification was calculated using LightCycler Software Version 4.5 (Roche Diagnostics) with the reaction’s efficiency correction. The results are shown as relative expression of the target gene relative to Gapdh or Hprt. The sequences of all primers are listed in ESM Table 2.
Protein isolation and western blots
Total proteins were isolated from cells as described by Goncz et al. [13]. For GLUT4 determination, plasma membrane fractions were prepared using the ProteoJET Membrane Protein Extraction Kit (Fermentas Life Sciences, St Leon-Rot, Germany). Contamination with cytoplasmatic and nuclear protein was excluded using western blots to detect β-actin. Ten micrograms of protein was resolved by SDS-PAGE and blotted onto nitrocellulose membranes [13]. After exposure to primary antibody overnight at 4°C, membranes were extensively washed and incubated with a secondary antibody for 1 h at room temperature. Signals were detected with an ECL kit (GE Healthcare Europe, Freiburg, Germany) [13]. Signal intensity was quantified with Quantity One 1-D Analysis Software (Bio-Rad Laboratories, Munich, Germany). The results are shown as relative (percentage) levels vs β-actin. In the case of GLUT4, the results are expressed relative to membrane protein concentration.
Glucose uptake
3T3-L1 adipocytes
Adipocytes in 24-well plates were cultured overnight in serum-free DMEM. Cells were then washed in PBS and incubated in glucose-free KRBH (0.1% fatty acid-free BSA) for 15 min. Cells were washed with PBS and incubated in KRBH (0.1% BSA) with test peptides. Subsequently, 18.5 kBq of 2-deoxy-d-[2,6-3H]glucose and 0.1 mmol/l 2-deoxyglucose were added. After 5 min, the reaction was terminated using 20 μmol/l of cytochalasin B. The medium was removed and the cells were washed with ice-cold PBS. Cells were lysed for 10 min in 0.1% SDS. Aliquots of cell lysates were used for liquid scintillation and for protein concentration determination by BCA protein assay (Thermo Scientific, Rockford, IL, USA). Results were normalised for protein concentration and presented as percentage of basal (uptake in the absence of test peptides).
Rat adipocytes
Glucose transport was studied in isolated rat adipocytes using 2-deoxy-d-[1-3H]glucose [23]. Briefly, adipocytes were preincubated in glucose-free KRBH containing test peptides for 30 min at 37°C in a shaking water-bath. Subsequently, 18.5 kBq of 2-deoxy-d-[1-3H]glucose and 0.5 mmol/l 2-deoxyglucose were added to adipocytes at 15 s intervals, and incubation was continued for 4 min The reaction was terminated using ice-cold 0.25 mmol/l phloretin within this same time period. The cell suspension was transferred to fresh tubes prefilled with silicone oil and centrifuged at 6,000×g for 1 min Afterwards, pellets were transferred to the scintillation vials and radioactivity was measured using a β-counter.
Phosphatidylinositol (3,4,5)-trisphosphate mass assay
Phosphatidylinositol (3,4,5)-trisphosphate (PIP3) was measured using the PIP3 Mass Elisa Kit (Echelon Biosciences, Salt Lake City, UT, USA).
Overnight serum-starved 3T3-L1 adipocytes, cultured in six-well plates, were incubated in serum-free DMEM containing test peptides for 5 min and the reaction was terminated with ice-cold PBS. Acidic lipids were extracted as described by Gray et al. [24], and resuspended in 60 μl of PBS-Tween-20 and 3% Protein Stabilizer buffer (provided with the ELISA kit). The extract was sonicated. PIP3 was measured in the fraction, according to the manufacturer’s protocol. Absorbance values were read at 450 nm and the amount of PIP3 was estimated based on the values in the standard curve that was generated.
Immunofluorescence
3T3-L1 adipocytes grown on coverslips were fixed with 4% paraformaldehyde for 20 min, washed with PBS for 10 min, and blocked with 2% BSA in PBS for 30 min After washing, cells were incubated with anti-GLUT4 diluted 1:100 in 1% BSA in PBS overnight at 4°C. Cells were washed and incubated for 60 min at room temperature with anti-rabbit cyanine dye 3 (Cy3) diluted 1:150 in 1% BSA in PBS. After washing with PBS, coverslips were mounted in Fluoromount G (Electron Microscopy Sciences, Fort Washington, PA, USA). Images were visualised with an FV-500 laser scanning confocal microscope (Olympus, Hamburg, Germany) at ×40 magnification.
Oil red O staining
Overnight serum starved adipocytes cultured in 12-well plates were incubated in serum-free DMEM containing test peptides. After 48 h, cells were washed with PBS and fixed with 10% formalin for 10 min. The cells were then washed with 60% isopropanol and air-dried. Cells were stained with Oil Red O (ORO) for 10 min. Excess ORO was washed away with water. Images were taken with an Axiophot microscope (Zeiss, Oberkochen, Germany) using a digital camera (AxioCam HRc; Zeiss) at ×20 magnification. For quantification, ORO was eluted from the cells using 100% isopropanol and the absorbance of the eluate was determined at 500 nm wavelength against isopropanol (blank).
Triacylglycerol content
Adipocytes were prepared using the same method as for the ORO assay. After exposure to test peptides, cells were washed with PBS. Cells were then scraped and sonicated in a triacylgycerol assay buffer containing 50 mmol/l Tris–HCl (pH 7.4) and 1 mmol/l EDTA. Triacylgycerol was determined using a colorimetric triacylglycerol kit (Cypress Diagnostics, Langdorp, Belgium).
Lipogenesis
Lipogenesis was evaluated based on the incorporation of [U-14C]glucose into NEFA as described above [25]. Adipocytes (1 × 106 cells/ml) were incubated in KRBH (3 mmol/l glucose, 3% NEFA-free BSA) with or without test peptides and with 18.5 kBq [U-14C]glucose for 2 h at 37°C in a shaking water bath. The assay was terminated by adding Dole’s extraction mixture [26]. Samples were shaken, mixed with H2O and heptan, and shaken again. The upper phase containing lipids was transferred to scintillation liquid for counting of incorporated radioactivity using a β-counter. The results are expressed as percentage of basal (set to 100%) incorporation of 14C into NEFA. For the ex vivo study, adipocytes were isolated from rats treated for 4 weeks with OXA (50 pmol/h), as described above. Basal conversion of glucose into NEFA was quantified (cpm/1 × 106 cells).
Lipolysis
Overnight serum-starved 3T3-L1 cells were incubated in serum-free DMEM (0.1% BSA) with test substances for 24 h. Medium was collected and then centrifuged at 800×g for 10 min. Free glycerol (index of lipolysis) was measured from the supernatant fraction using a Free Glycerol Determination Kit (Sigma-Aldrich).
Pparγ siRNA transfection
Adipocytes were transfected with 200 nmol/l mouse Pparγ or with non-targeting small interfering siRNA (siRNA) (ON-Target plus SMART; Dharmacon, Lafayette, CO, USA) using a Deliver X Plus siRNA Transfection Kit (Panomics, Santa Clara, CA, USA) as described in the manufacturer’s instructions. The efficiency of gene silencing was evaluated by real-time PCR and western blotting.
Adiponectin secretion and plasma concentrations of adiponectin and OXA
Adipocytes were incubated overnight in serum-free DMEM (0.1% BSA). After repeated washing with PBS, cells were incubated in serum-free DMEM with test agents. Medium was collected and then centrifuged for 10 min at 800×g. The supernatant fraction was stored at −70°C. Plasma adiponectin concentration was measured in chronically OXA-treated rats, which were then starved overnight. Adiponectin concentration was measured using a mouse/rat RIA test for adiponectin (Linco Research, St Charles, MO, USA). OXA was measured using an orexin A ELISA kit provided by Phoenix Europe (Karslruhe, Germany).
Statistical analysis
Results are presented as mean ± SEM. Means were obtained from five or six independent experiments performed at least in duplicate. Statistical analysis was performed using unpaired Student’s t test (two-tailed distribution), and statistical significance was accepted at p < 0.05.
Results
Levels of OXR1 and OXR2 in adipocytes
OXR1 and OXR2 were produced in 3T3-L1 fibroblasts, mature 3T3-L1 adipocytes and in primary rat adipocytes (Fig. 1a, b). Notably, Oxr1 RNA expression increased during the differentiation process of 3T3-L1 fibroblasts into adipocytes, whereas the expression of Oxr2 remained unchanged (Fig. 1a). We confirmed the production of both receptor subtypes in adipocytes on protein extracts by western blotting (Fig. 1c).

Detection of Oxr1 and Oxr2 expression in adipocytes. a Detection of Oxr1 and Oxr2 by quantitative PCR in 3T3-L1 adipocytes. Note the time-dependent changes in Oxr1 RNA expression during the differentiation process of 3T3-L1 cells from fibroblasts into mature adipocytes. Day 0 denotes onset of the differentiation process, days 4 and 7 are the fourth and the seventh days after completion of the differentiation of 3T3-L1 cells into mature adipocytes. White bars, Oxr1; black bars, Oxr2. Results show relative expression of Oxr1 and Oxr2 normalised to Gapdh. *p < 0.05 vs corresponding controls, Student’s t test. Data are derived from four independent experiments. b Detection of Oxr1 and Oxr2 RNA by quantitative PCR in primary rat adipocytes, freshly isolated and pooled from three adult male rats. Each experiment was performed in quadruplicates and the results were repeated once. c Qualitative detection of OXR1 (55 kDa) and OXR2 (38 kDa) proteins in isolated primary rat adipocytes and in 3T3-L1 adipocytes by western blotting. Results were repeated once using different batches of 3T3-L1 cells and different preparations of rat adipocytes (each experiment was performed in quadruplicate)
OXA stimulates active glucose uptake
Since glucose provides a relevant source for lipid synthesis, we characterised the effect of OXA on active glucose uptake. OXA (100 nmol/l) time-dependently increased glucose uptake in 3T3-L1 adipocytes, reaching the maximum after 30 min (Fig. 2a). Concentration–response analysis at 30 min revealed that maximal stimulation was induced by 100 nmol/l OXA (Fig. 2b).

Effects of OXA on glucose uptake and PI3K. a Time-dependent stimulation of glucose uptake in 3T3-L1 adipocytes by 100 nmol/l OXA. b Concentration-dependent increase in glucose uptake by 3T3-L1 adipocytes exposed to OXA for 30 min c Stimulation of glucose uptake by 100 nmol/l OXA, 10 nmol/l insulin and the combination of both in isolated primary rat adipocytes after 30 min of incubation. Ins, insulin. d Inhibition of 100 nmol/l OXA- and 10 nmol/l insulin-stimulated glucose uptake by the PI3K inhibitor LY294002 in 3T3-L1 adipocytes. Results show the cellular content of 2-deoxy-[3H]glucose (normalised vs protein content) taken up in the presence of OXA (at the indicated concentrations) or 10 nmol/l insulin or in the presence of both 100 nmol/l OXA and 10 nmol/l insulin. The standard concentration was 100 nmol/l for OXA and 10 nmol/l for insulin unless otherwise stated. *p < 0.05, **p < 0.01, ***p < 0.001 vs corresponding controls (vehicle-treated cells) using Student’s t test. Data are derived from four or five independent experiments. e Stimulation of cellular PIP3 production in 3T3-L1 adipocytes exposed for 5 min to 100 nmol/l OXA or treated with 10 nmol/l insulin. Results are expressed as nmol/well and the data are derived from four experiments. Ins, insulin. **p < 0.01, ***p < 0.001 vs controls (vehicle-treated 3T3-L1 cells), Student’s t test
OXA stimulated glucose uptake in isolated rat adipocytes, albeit less potently than insulin, tested at the most effective concentration of 10 nmol/l (Fig. 2c). OXA further enhanced insulin-induced glucose uptake (Fig. 2c).
PI3K plays a role in regulating active glucose uptake through activation of GLUT4. We therefore incubated 3T3-L1 adipocytes and isolated rat adipocytes for 30 min with OXA or insulin (positive control) in the presence of the PI3K inhibitor LY294002. LY294002 inhibited OXA- and insulin-stimulated glucose uptake in 3T3-L1 adipocytes (Fig. 2d) and in isolated rat adipocytes (data not shown). To confirm the role of PI3K, we measured PIP3 production in 3T3-L1 cells exposed for 5 min to OXA or insulin. Both peptides increased cellular PIP3 content (Fig. 2e). In agreement with this, we detected a time-dependent increase in phosphorylation of the p85 subunit of PI3K in 3T3-L1 cells treated with OXA and insulin (ESM Fig. 2a, b). OXA also increased the phosphorylation of PI3K downstream effector proteins pdk1 and protein kinase B (AKT) (ESM Fig. 2c–e). These data suggest that OXA increases glucose uptake via PI3K.
OXA increases the GLUT4 plasma membrane translocation via PI3K
Active glucose uptake by adipocytes is regulated via GLUT4, which translocates from the cytoplasm into the plasma membrane compartment upon activation. The level of GLUT4 protein in the plasma membrane of 3T3-L1 adipocytes increased in response to treatment with OXA or insulin (Fig. 3a). Using immunofluorescence, we detected GLUT4 translocation from the cytoplasm into the cell periphery (Fig. 3b). The PI3K inhibitor LY294002 prevented OXA and insulin from inducing GLUT4 translocation (Fig. 3c), indicating that OXA PI3K-dependently increases glucose uptake and the translocation of GLUT4.

PI3K-dependent translocation of GLUT4 from cytoplasm into plasma membrane in adipocytes exposed to OXA. a Translocation of GLUT4 into the plasma membrane after exposing 3T3-L1 adipocytes to 100 nmol/l OXA or 10 nmol/l insulin for 30 min. Bar chart shows quantification of GLUT4 protein isolated from plasma membranes and detected by western blots. Gels show representative western blot showing GLUT4 proteins in the plasma membrane fraction, isolated from adipocytes. **p < 0.01, ***p < 0.001 vs non-stimulated controls, Student’s t test. Data are derived from three independent experiments. b Visualisation of GLUT4 translocation from the cytoplasm into the plasma membrane of 3T3-L1 adipocytes exposed for 30 min to 100 nmol/l OXA or 10 nmol/l insulin by immunofluorescence. Images were acquired by laser scanning confocal microscopy at ×40 magnification. c Inhibition of OXA-stimulated GLUT4 translocation into plasma membrane by PI3K inhibitor LY294002. Results demonstrate western blotting performed on proteins isolated from membranes of 3T3-L1 adipocytes exposed to vehicle or 100 nmol/l OXA in the presence or absence of LY294002. Data are derived from two independent experiments. *p < 0.05, **p < 0.01 vs non-stimulated controls, Student’s t test. d Time-dependent stimulation of AS160 phosphorylation by 100 nmol/l OXA and by 10 nmol/l insulin. AS160 is the AKT substrate regulating GLUT4 translocation. Representative western blots for proteins isolated from 3T3-L1 adipocytes exposed to vehicle, OXA or insulin. Bar graph shows the quantitative analysis of western blot images. Data are derived from three independent experiments. **p < 0.01, ***p < 0.001 vs non-stimulated controls (time 0), Student’s t test
It is known that insulin stimulates GLUT4 translocation via a PI3K/AKT-dependent pathway. AS160 is one of the AKT substrates that, in the phosphorylated state, stimulates GLUT4 translocation. Consistent with this notion, we observed increased phosphorylation of AS160 in insulin- and OXA-treated 3T3-L1 adipocytes (Fig. 3d).
OXA stimulates triacylglycerol synthesis via PI3K
Since OXA increased glucose uptake, we expected an increased triacylglycerol content. As predicted, OXA (concentration-dependently) increased triacylglycerol content in 3T3-L1 adipocytes after 48 h (Fig. 4a) and potentiated insulin-stimulated triacylglycerol accumulation (Fig. 4b–f). The increased lipid content in both OXA and OXA/insulin-treated adipocytes resulted from the stimulation of lipogenesis, detected by the incorporation of glucose into NEFA (Fig. 4g). The incorporation of glucose into NEFA in adipocytes isolated from rats treated for 4 weeks with OXA was increased approximately twofold compared with that in adipocytes isolated from vehicle-treated rats (Fig. 4h). This result further supports the in vitro anabolic activity of OXA in adipocytes.

PI3K-dependent regulation of triacylglycerol accumulation by OXA. a Quantification of triacylglycerol content in 3T3-L1 adipocytes exposed to OXA (1–100 nmol/l) for 48 h using oil red O staining. After staining of adipocytes with oil red O, the dye was eluted and the optical density (OD) at wavelength 500 nm was determined with a spectrophotometer. Results are expressed as OD at 500 nm. b–f Effects of 100 nmol/l OXA, 10 nmol/l insulin and the combination of both peptides on lipid accumulation in 3T3-L1 adipocytes after 48 h of treatment. b Quantification of ORO staining. c–f Corresponding representative images of ORO-stained sections at ×20 magnification: (c) vehicle, (d) 100 nmol/l OXA, (e) 10 nmol/l insulin, (f) 100 nmol/l OXA together with 10 nmol/l insulin. Ins, insulin. g Conversion of glucose into NEFA in isolated primary rat adipocytes in response 100 nmol/l OXA, 10 nmol/l insulin or both for 2 h. Results are shown as percentage of basal. Basal (set to 100%) is the conversion of glucose into NEFA in adipocytes in the presence of vehicle and without test agents. Ins, insulin. h Ex vivo conversion of glucose into NEFA in adipocytes isolated from overnight-fasted rats treated for 4 weeks with OXA (50 pmol/h, i.p.). Adipocytes freshly isolated from rats that had been chronically treated either with vehicle (0.9% NaCl) or OXA were incubated with 3 mmol/l glucose and 18.5 kBq [U-14C]glucose with no additional test peptides. NEFA content was measured after 120 min Results are expressed as cpm/106 cells. **p < 0.01 vs corresponding control cells derived from rats treated with vehicle, Student’s t test. i Effect of PI3K inhibitor LY294002 (10 μmol/l) on triacylglycerol accumulation in 3T3-L1 adipocytes treated with 100 nmol/l OXA or 10 nmol/l insulin (positive control) for 24 h. Triacylglycerol content was determined biochemically and normalised to protein concentration. j Inhibition of 100 nmol/l OXA- and 10 nmol/l insulin-stimulated conversion of glucose into NEFA by LY294002 in isolated primary rat adipocytes. Results are shown as percentages of basal. Basal (100%) was the conversion of glucose into NEFA in adipocytes without test agents. k Determination of glycerol release (index of lipolysis) from isolated 3T3 L1 exposed to 100 nmol/l OXA for 24 h. Results are shown as concentrations of free glycerol in incubation buffer. Free glycerol concentration is normalised to total protein concentration. Isoprenaline (1 μmol/l) and insulin (10 nmol/l) were used as known lipolytic and antilipolytic controls. Isopren, isoprenaline; Ins, insulin. Inhibition of Hsl mRNA expression in 3T3-L1 cells exposed for 24 h to either 10 nmol/l insulin or 100 nmol/l OXA. Expression of Hsl was normalised to Hprt. Ins, insulin. Unless otherwise stated, all data in Fig. 4 are derived from three to six independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs corresponding controls (as indicated). Student’s t test
We further elucidated the mechanism of OXA action by exposing adipocytes to LY294002. This PI3K inhibitor blocked OXA- and insulin-induced triacylglycerol accumulation (Fig. 4i) and the conversion of glucose into NEFA in rat adipocytes (Fig. 4j).
It is known that the net effect on lipid accumulation results from lipogenesis and lipolysis. We therefore measured the effects of OXA on lipolysis, indexed by the amount of released glycerol. OXA and insulin inhibited lipolysis, whereas isoprenaline (positive control) stimulated lipolysis from 3T3-L1 adipocytes (Fig. 4k). OXA reduced the mRNA expression of Hsl without affecting the expression of adipose triacylglycerol lipase and perilipin (Fig. 4l and data not shown). These data suggest that OXA PI3K-dependently stimulates lipid synthesis and reduces lipolysis in adipocytes.
OXA stimulates triacylglycerol accumulation via PPARγ-dependent mechanism
Earlier studies have shown that triacylglycerol synthesis is regulated by PPARγ-dependent mechanisms. PPARγ2 is the major PPARγ isoform produced in adipocytes. OXA concentration-dependently upregulated Pparγ2 mRNA expression in rat adipocytes and in 3T3-L1 adipocytes (Fig. 5a, b). In addition, OXA potentiated insulin-stimulated Pparγ2 expression (Fig. 5c). The effects of pharmacological inhibition of PPARγ activity on OXA-induced lipogenesis were tested. OXA increased triacylglycerol content in 3T3-L1 adipocytes, which was prevented by the PPARγ antagonist BADGE (Fig. 5d). The level of PPARγ2 in 3T3-L1 cells was reduced by approximately 70% using siRNA (ESM Fig. 3), which resulted in a loss of OXA-induced triacylglycerol accumulation (Fig. 5e). An nTZD PPARγ agonist (used as positive control) stimulated triacylglycerol accumulation (Fig. 5f). The effect was lost in 3T3-L1 adipocytes treated with Pparγ siRNA (Fig. 5f), indicating that OXA increases triacylglycerol accumulation in 3T3-L1 adipocytes via a PPARγ-dependent mechanism.

Effect of OXA on Pparγ2 expression in adipocytes. a Concentration-dependent stimulation of Pparγ2 expression by OXA in isolated rat adipocytes. The graph illustrates results obtained by real-time PCR using total RNA isolated from rat primary adipocytes exposed to 1–100 nmol/l OXA for 8 h. Relative expression of Pparγ2 is normalised to the housekeeping gene Hprt. b Concentration-dependent stimulation of Pparγ2 expression in 3T3-L1 adipocytes treated for 24 h with OXA (1–100 nmol/l). Relative expression of Pparγ2 is normalised to Hprt. c Effects of 100 nmol/l OXA, 10 nmol/l insulin and the combination of both peptides on Pparγ2 expression in 3T3-L1 adipocytes incubated with test peptides for 24 h. Ins, insulin. d Effects of the PPARγ inhibitor BADGE (50 μmol/l) on triacylglycerol accumulation in 3T3-L1 adipocytes treated with 100 nmol/l OXA for 24 h. Triacylglycerol content was determined biochemically and is normalised to protein concentration. White bars, cells incubated without BADGE; black bars, BADGE-treated cells. e Effects of 100 nmol/l OXA on triacylglycerol accumulation in 3T3-L1 adipocytes treated with Pparγ siRNA. For all experiments performed on cells treated with Pparγ siRNA, cells exposed to scrambled non-targeting siRNA served as controls. White bars, vehicle-treated cells; black bars, OXA-treated cells. f Effects of an nTZD PPARγ agonist (10 μmol/l) on triacylglycerol accumulation in 3T3-L1 cells treated with the Pparγ siRNA. Triacylglycerol content is normalised to protein concentration. White bars, vehicle-treated cells; black bars, nTZD-treated cells. All data in Fig.5 are derived from three to six independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs corresponding controls (as indicated), Student’s t test
OXA stimulates adiponectin secretion and gene expression via PPARγ-dependent mechanism
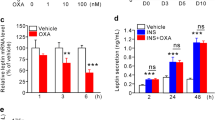
PPARγ-agonists are known to stimulate adiponectin secretion. Since OXA increased the level of PPARγ2, we tested the influence of OXA on adiponectin secretion. OXA concentration-dependently increased adiponectin secretion from 3T3-L1 cells but failed to potentiate nTZD-stimulated adiponectin secretion (Fig. 6a). In addition, OXA increased cellular adiponectin protein content (Fig. 6b, c). This effect was lost in the presence of BADGE (Fig. 6b, c) and in adipocytes with attenuated PPARγ levels (Fig. 6d, e). Plasma adiponectin levels measured at 4 week intervals in OXA-treated rats fed chow or HFD were increased compared with vehicle-treated rats (Fig. 6f). Taken together, these results indicate that OXA stimulates adiponectin secretion and gene expression in adipocytes by a PPARγ-dependent mechanism.

Role of PPARγ in OXA-regulated adiponectin secretion. a Concentration–response evaluation of adiponectin secretion from 3T3-L1 adipocytes incubated with OXA for 24 h. Stimulation of adiponectin secretion by a non-TZD PPARγ agonist (10 μmol/l) alone and in combination with 10 nmol/l OXA. Results are concentration of released adiponectin (ng/ml). Black bars, nTZD-treated cells; white bars, without nTZD. *p < 0.05, ***p < 0.001 vs non-treated controls, Student’s t test. Data are derived from six independent experiments. b, c Effect of OXA (100 nmol/l) on adiponectin protein accumulation in 3T3-L1 adipocytes in the presence or absence of the PPARγ antagonist BADGE (50 μmol/l), determined by quantitative western blot analysis. b Quantification of western blot signals expressed as the ratio of adiponectin to β-actin signal intensity (arbitrary units). Black bars, BAGDE-treated cells; white bars, without BADGE. c Representative western blot showing adiponectin protein band and the band for the housekeeping gene product for β-actin. d, e Effects of 100 nmol/l OXA on adiponectin protein expression in adipocytes treated with Pparg siRNA, determined by western blotting (quantitative analysis). Adiponectin protein levels in cells treated with scrambled non-targeting siRNA were used as controls. Black bars, OXA-treated cells; white bars, without OXA. d Quantification of western blots; e representative western blots with protein signals for adiponectin and β-actin. *p < 0.05, **p < 0.01, ***p < 0.001 vs non-treated controls, Student’s t test. Data are derived from three to six independent experiments. f Adiponectin plasma concentration measured in adult Wistar rats fed either a standard chow diet or HFD after continuous application of OXA at 50 pmol/h for 4 weeks using osmotic pumps. Rats receiving vehicle (0.9% NaCl) were used as controls. Data are means of five or six animals per group. *p < 0.05, **p < 0.01 vs corresponding controls, Student’s t test
Discussion
The major novel finding of our study is that OXA stimulates lipid accumulation in isolated adipocytes. The anabolic activity of OXA is based on the PI3K- and PPARγ-dependent stimulation of lipogenesis and the inhibition of lipolysis. OXA PI3K-dependently increases membrane translocation of GLUT4 and the active uptake of glucose, which is then converted to triacylglycerol. In addition, we show that OXA stimulates adiponectin levels via PPARγ.
A previous study using Oxr1-transfected HEK293 cells showed that OXA can increase glucose uptake via hypoxia-inducible factor 1-alpha (HIF1α)-dependent stimulation of GLUT1 production [19]. In adipocytes, GLUT4 is the major glucose transporter, which PI3K-dependently translocates into the plasma membrane upon exposure to insulin [27]. We show that OXA PI3K-dependently induces GLUT4 translocation and active glucose uptake in adipocytes. In addition, OXA stimulates the phosphorylation of the AKT substrate AS160, which facilitates GLUT4 translocation. This observation suggests that OXA, like insulin, stimulates GLUT4 via PI3K/AKT.
Increased glucose uptake by adipocytes in response to OXA treatment may possibly explain the mechanism by which OXA lowers blood glucose levels in non-diabetic and diabetic animals without changes in insulin concentration [15].
Orexin acutely stimulates food intake [1]. However, animals overproducing orexin are lean because of increased energy expenditure [4]. The increased energy expenditure possibly results from increased release of stress hormones and/or increased levels and/or sensitivity to leptin [4]. Leptin can increase energy expenditure by decreasing NPY/AGRP expression and or activating pro-opiomelanocortin (POMC) neurons [28]. In our study, rats treated with OXA for 4 weeks did not show any significant changes in body weight, which may be attributable to the limited duration of the study or to increased energy expenditure. However, energy expenditure was not assessed in our study.
In contrast to the antiobesity effects reported in animals overexpressing orexin or its receptors, OXA increased triacylglycerol accumulation in isolated adipocytes, which is consistent with the results of a previous study in isolated human adipocytes [18]. The lipogenic activity of OXA is further substantiated by ex vivo observation using adipocytes isolated from rats treated chronically with OXA for 4 weeks. These adipocytes displayed increased conversion of glucose to NEFA. PI3K activation is known to enhance lipid accumulation in 3T3-L1 adipocytes [29]. In agreement with this, we demonstrate that PI3K inhibition prevents OXA-induced triacylglycerol accumulation, as well as the conversion of glucose to NEFA. In addition, OXA diminished lipolysis in the current study. Inhibition of HSL expression appears to be a mechanism of antilipolytic activity of OXA, as previously demonstrated [18] and confirmed in our current study.
Earlier work demonstrated that OXA stimulates the proliferation of 3T3-L1 pre-adipocytes [17]. The differentiation of adipocytes critically depends upon PPARγ [30, 31]. Increased PPARγ2 expression stimulates the proliferation of adipocytes and the lipid accumulation [32, 33]. The PPARγ2 isoform is restricted to and highly abundant in adipocytes [33, 34]. Animals lacking Pparγ2 are considered a model of fat-specific Pparγ inactivation [35]. These mice have markedly reduced white adipose tissue content and impaired insulin sensitivity [35]. In a previous study, OXA increased PPARγ2 protein levels in adipocytes [18], possibly representing the mechanism of lipogenic activity of OXA. Evidence supporting the role of PPARγ in OXA-stimulated triacylglycerol is substantiated by the following results. The pharmacological blockade of PPARγ by the antagonist BADGE, as well as the attenuation of Pparγ expression by siRNA, prevented OXA from stimulating triacylglycerol synthesis. The same observation was made using a known nTZD PPARγ agonist, which lost its lipogenic activity in Pparγ siRNA-treated adipocytes. These results suggest that OXA stimulates lipogenesis via PPARγ. In addition, OXA potentiates the insulin-induced stimulation of triacylglycerol synthesis, and is presumably also PPARγ-dependent. Although the mechanisms through which OXA stimulates Pparγ expression are unknown, we hypothesise that the arachidonic acid (AA)/prostaglandin pathway may represent a link. Two recent studies showed that activation of OXR1 by OXA strongly increased the release of AA, an effect mediated through phospholipase A2 [36, 37]. The exact mechanisms of this activation are not yet known; however, Ca2+ seems to play a role in this process. Ca2+ is a potent stimulus of AA release. Importantly, OXA is known to increase intracellular Ca2+, as a result of activation of different transient receptor potential channels [37]. AA is a precursor for the synthesis of prostaglandins, such as PGJ2. PGJ2 metabolites can act as endogenous PPARγ ligands, which are able to upregulate the production, transcriptional activity and DNA binding activity of PPARγ [36, 38].
PPARγ plays a key role in regulating the synthesis and release of adipokines. Recent in vivo studies have demonstrated that orexin can increase insulin sensitivity [4]. Although some of the anti-obesity effects of OXA in vivo possibly result from enhanced sensitivity to leptin, the mechanism of OXA-improved insulin sensitivity is not entirely known. Adiponectin is able to reduce peripheral insulin resistance in type 2 diabetes mellitus [39]. The secretion of adiponectin from fat cells can be stimulated by PPARγ activators [40]. In our study, OXA stimulated the secretion of adiponectin. The effect was lost in Pparγ siRNA-treated adipocytes. In addition, adiponectin plasma levels were increased in chronically OXA-treated rats. Although we did not show this in our study, we speculate that adiponectin, released in response to OXA, could contribute to the previously reported improvement of insulin sensitivity in mice [4].
In summary, we demonstrate that OXA stimulates glucose uptake via GLUT4 and triacylglycerol synthesis in isolated adipocytes through a PI3K-dependent mechanism. OXA increases PPARγ levels, with the concomitant stimulation of triacylglycerol synthesis and adiponectin secretion. We hypothesise that OXA may play a role in pathophysiology of obesity and type 2 diabetes mellitus.
Abbreviations
- AKT/PKB:
-
Protein kinase B
- BADGE:
-
Bisphenol A diglycidyl ether
- HFD:
-
High-fat diet
- HPRT:
-
Hypoxanthine-guanine phosphoribosyltransferase
- HSL:
-
Hormone-sensitive lipase
- KRBH:
-
Krebs–Ringer bicarbonate HEPES buffer
- nTZD:
-
Non-thiazolidinedione PPARγ activator
- ORO:
-
Oil Red O
- OXA:
-
Orexin A
- OXR1:
-
Orexin receptor subtype 1
- OXR2:
-
Orexin receptor subtype 2
- PI3K:
-
Phosphoinositide 3-kinase
- PIP3:
-
Phosphatidylinositol (3,4,5)-trisphosphate
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- siRNA:
-
Small interfering RNA
References
Sakurai T, Amemiya A, Ishii M et al (1998) Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92:573–585
Hara J, Beuckmann CT, Nambu T et al (2001) Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30:345–354
Hara J, Yanagisawa M, Sakurai T (2005) Difference in obesity phenotype between orexin-knockout mice and orexin neuron-deficient mice with same genetic background and environmental conditions. Neurosci Lett 380:239–242
Funato H, Tsai AL, Willie JT et al (2009) Enhanced orexin receptor-2 signaling prevents diet-induced obesity and improves leptin sensitivity. Cell Metab 9:64–76
Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E (2000) Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355:39–40
Nishino S, Ripley B, Overeem S et al (2001) Low cerebrospinal fluid hypocretin (Orexin) and altered energy homeostasis in human narcolepsy. Ann Neurol 50:381–388
Honda Y, Doi Y, Ninomiya R, Ninomiya C (1986) Increased frequency of non-insulin-dependent diabetes mellitus among narcoleptic patients. Sleep 9:254–259
Adam JA, Menheere PP, van Dielen FM, Soeters PB, Buurman WA, Greve JW (2002) Decreased plasma orexin-A levels in obese individuals. Int J Obes Relat Metab Disord 26:274–276
Nakabayashi M, Suzuki T, Takahashi K et al (2003) Orexin-A expression in human peripheral tissues. Mol Cell Endocrinol 205:43–50
Ehrstrom M, Naslund E, Levin F et al (2004) Pharmacokinetic profile of orexin A and effects on plasma insulin and glucagon in the rat. Regul Pept 119:209–212
Ouedraogo R, Naslund E, Kirchgessner AL (2003) Glucose regulates the release of orexin-a from the endocrine pancreas. Diabetes 52:111–117
Switonska MM, Kaczmarek P, Malendowicz LK, Nowak KW (2002) Orexins and adipoinsular axis function in the rat. Regul Pept 104:69–73
Goncz E, Strowski MZ, Grotzinger C et al (2008) Orexin-A inhibits glucagon secretion and gene expression through a Foxo1-dependent pathway. Endocrinology 149:1618–1626
Nowak KW, Strowski MZ, Switonska MM et al (2005) Evidence that orexins A and B stimulate insulin secretion from rat pancreatic islets via both receptor subtypes. Int J Mol Med 15:969–972
Tsuneki H, Sugihara Y, Honda R, Wada T, Sasaoka T, Kimura I (2002) Reduction of blood glucose level by orexins in fasting normal and streptozotocin-diabetic mice. Eur J Pharmacol 448:245–252
Tsuneki H, Murata S, Anzawa Y et al (2008) Age-related insulin resistance in hypothalamus and peripheral tissues of orexin knockout mice. Diabetologia 51:657–667
Zwirska-Korczala K, Mczyk-Sowa M, Sowa P et al (2007) Role of leptin, ghrelin, angiotensin II and orexins in 3T3 L1 preadipocyte cells proliferation and oxidative metabolism. J Physiol Pharmacol 58(Suppl 1):53–64
Digby JE, Chen J, Tang JY, Lehnert H, Matthews RN, Randeva HS (2006) Orexin receptor expression in human adipose tissue: effects of orexin-A and orexin-B. J Endocrinol 191:129–136
Sikder D, Kodadek T (2007) The neurohormone orexin stimulates hypoxia-inducible factor-1 activity. Genes Dev 21:2995–3005
Berger J, Tanen M, Elbrecht A et al (2001) Peroxisome proliferator-activated receptor-gamma ligands inhibit adipocyte 11beta-hydroxysteroid dehydrogenase type 1 expression and activity. J Biol Chem 276:12629–12635
Rodbell M (1964) Metabolism of isolated fat cells. I. Effects of hormones on glucose metabolism and lipolysis. J Biol Chem 239:375–380
Nugent C, Prins JB, Whitehead JP et al (2001) Potentiation of glucose uptake in 3T3-L1 adipocytes by PPAR gamma agonists is maintained in cells expressing a PPAR gamma dominant-negative mutant: evidence for selectivity in the downstream responses to PPAR gamma activation. Mol Endocrinol 15:1729–1738
Tang S, Le-Tien H, Goldstein BJ, Shin P, Lai R, Fantus IG (2001) Decreased in situ insulin receptor dephosphorylation in hyperglycemia-induced insulin resistance in rat adipocytes. Diabetes 50:83–90
Gray A, Olsson H, Batty IH, Priganica L, Peter DC (2003) Nonradioactive methods for the assay of phosphoinositide 3-kinases and phosphoinositide phosphatases and selective detection of signaling lipids in cell and tissue extracts. Anal Biochem 313:234–245
Szkudelski T, Szkudelska K, Nogowski L (2009) Effects of adenosine A1 receptor antagonism on lipogenesis and lipolysis in isolated rat adipocytes. Physiol Res 58:863–871
Dole VP, Meinertz H (1960) Microdetermination of long-chain fatty acids in plasma and tissues. J Biol Chem 235:2595–2599
Somwar R, Niu W, Kim DY et al (2001) Differential effects of phosphatidylinositol 3-kinase inhibition on intracellular signals regulating GLUT4 translocation and glucose transport. J Biol Chem 276:46079–46087
van de Wall E, Leshan R, Xu AW et al (2008) Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology 149:1773–1785
Xia X, Serrero G (1999) Inhibition of adipose differentiation by phosphatidylinositol 3-kinase inhibitors. J Cell Physiol 178:9–16
Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM (1994) mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev 8:1224–1234
Auwerx J, Schoonjans K, Fruchart JC, Staels B (1996) Regulation of triglyceride metabolism by PPARs: fibrates and thiazolidinediones have distinct effects. J Atheroscler Thromb 3:81–89
Rieusset J, Andreelli F, Auboeuf D et al (1999) Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes 48:699–705
Spiegelman BM (1998) PPAR-gamma: adipogenic regulator and thiazolidinedione receptor. Diabetes 47:507–514
Giusti V, Verdumo C, Suter M, Gaillard RC, Burckhardt P, Pralong F (2003) Expression of peroxisome proliferator-activated receptor-gamma1 and peroxisome proliferator-activated receptor-gamma2 in visceral and subcutaneous adipose tissue of obese women. Diabetes 52:1673–1676
Zhang J, Fu M, Cui T et al (2004) Selective disruption of PPARgamma 2 impairs the development of adipose tissue and insulin sensitivity. Proc Natl Acad Sci USA 101:10703–10708
Turunen PM, Ekholm ME, Somerharju P, Kukkonen JP (2010) Arachidonic acid release mediated by OX1 orexin receptors. Br J Pharmacol 159:212–221
Peltonen HM, Magga JM, Bart G et al (2009) Involvement of TRPC3 channels in calcium oscillations mediated by OX(1) orexin receptors. Biochem Biophys Res Commun 385:408–412
Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM (1995) A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83:813–819
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K (2006) Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 116:1784–1792
Combs TP, Wagner JA, Berger J et al (2002) Induction of adipocyte complement-related protein of 30 kilodaltons by PPARgamma agonists: a potential mechanism of insulin sensitization. Endocrinology 143:998–1007
Acknowledgements
The study was supported by the DFG grant STR558 to M. Z. Strowski and the Albert Renold Fellowship of the EASD to P. Pietrzak. nTZD was a gift from Merck Research Laboratories. The results of the study form part of the PhD thesis of M. Skrzypski. Linguistic advice from E. Zach is greatly acknowledged.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Fig. 1
a Plasma OXA concentration in rats treated continuously for 4 weeks with OXA (50 pmol/h), vehicle (0.9% NaCl) and fed either chow diet or HFD for 5 weeks. Animals with inserted osmotic pumps filled with vehicle or OXA were killed at the end of the study and the plasma concentration of OXA (picograms per nanolitre) was determined by ELISA. Data are mean ± SEM for five or six animals. Note the decreased endogenous plasma OXA concentration in HFD-treated rats compared with chow-fed rats. *p < 0.05 using Student’s t test. b Body weights of rats fed chow diet or HFD and treated continuously (i.p.) for 4 weeks with either vehicle or OXA. Data are mean ± SEM for five or six animals (PDF 104 kb)
ESM Fig. 2
a Stimulation of phosphorylation of p85 subunit of PI3K in 3T3-L1 adipocytes exposed to 100 nmol/l OXA for the indicated time periods. b Stimulation of phosphorylation of p85 subunit of PI3K in 3T3-L1 adipocytes exposed to 10 nmol/l insulin for 5 min. Western blot signals were quantified as described in Methods. Results are expressed in arbitrary units (signal intensity of p-p85 normalised to p85). The experiment was repeated twice and the mean ± SEM was calculated. **p < 0.01 vs vehicle-treated cells (time 0), Student’s t test. c Time-dependent stimulation of PDK1 phosphorylation in 3T3-L1 adipocytes by 100 nmol/l OXA. Images show representative Western blot signals. The experiment was performed twice. The intensity of p-PDK1 was quantified against total PDK1 and the results are shown as bar graphs representing the mean ± SEM. **p < 0.01, ***p < 0.001 vs vehicle-treated cells (time 0), Student’s t test. d Time-dependent stimulation of AKT phosphorylation at tyrosine by 100 nmol/l OXA. e Time-dependent stimulation of AKT phosphorylation at serine by 100 nmol/l OXA. Images show representative Western blot signals. The experiment was performed twice. The intensity of pAKT signal was quantified against total AKT and the results are shown as bar graphs, representing the mean ± SEM. **p < 0.01, ***p < 0.001 vs vehicle-treated cells (time 0), Student’s t test (PDF 320 kb)
ESM Fig. 3
Quantification of PPARγ2 proteins in 3T3-L1 cells treated with Pparγ siRNA for 72 h. Western blot signals were quantified as described in Methods. Results are expressed in arbitrary units (signal intensity of PPARγ2 normalised to β-actin). The experiment was performed twice and the mean ± SEM was calculated. ***p < 0.001 vs non-targeting siRNA-treated cells, Student’s t test (PDF 91 kb)
ESM Table 1
(PDF 35.2 kb)
ESM Table 2
(PDF 117 kb)
Rights and permissions
About this article
Cite this article
Skrzypski, M., T. Le, T., Kaczmarek, P. et al. Orexin A stimulates glucose uptake, lipid accumulation and adiponectin secretion from 3T3-L1 adipocytes and isolated primary rat adipocytes. Diabetologia 54, 1841–1852 (2011). https://doi.org/10.1007/s00125-011-2152-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-011-2152-2