
Abstract
Aims/hypothesis
Substantial evidence suggests a link between elevated inflammation and development of insulin resistance. Toll-like receptor 2 (TLR2) recognises a large number of lipid-containing molecules and transduces inflammatory signalling in a variety of cell types, including insulin-responsive cells. Considering the contribution of the fatty acid composition in TLR2-depedent signalling, we hypothesised that the inflammatory signals transduced by TLR2 contribute to insulin resistance.
Methods
Mice deficient in TLR2 were used to investigate the in vivo roles of TLR2 in initiating and maintaining inflammation-associated insulin resistance and energy homeostasis.
Results
We first recapitulated the observation with elevated expression of TLR2 and inflammatory cytokines in white adipose tissue and liver of ob/ob mice. Aged or high-fat-fed TLR2-deficient mice were protected from obesity and adipocyte hypertrophy compared with wild-type mice. Moreover, mice lacking TLR2 exhibited improved glucose tolerance and insulin sensitivity regardless of feeding them regular chow or a high-fat diet. This is accompanied by reductions in expression of inflammatory cytokines and activation of extracellular signal-regulated kinase (ERK) in a liver-specific manner. The attenuated hepatic inflammatory cytokine expression and related signalling are correlated with increased insulin action specifically in the liver in TLR2-deficient mice, reflected by increased insulin-stimulated protein kinase B (Akt) phosphorylation and IRS1 tyrosine phosphorylation and increased insulin-suppressed hepatocyte glucose production.
Conclusions/interpretation
The absence of TLR2 attenuates local inflammatory cytokine expression and related signalling and increases insulin action specifically in the liver. Thus, our work has identified TLR2 as a key mediator of hepatic inflammation-related signalling and insulin resistance.
Similar content being viewed by others


Introduction
Insulin resistance is the chief abnormality present in metabolic syndrome. Increasing evidence suggests causative links between inflammation and development of insulin resistance [1, 2]. Thus, the inflammatory nuclear factor kappa B (NF-κB) signalling pathway is activated and pro-inflammatory cytokines are produced in the white adipose tissue (WAT) and livers of insulin-resistant animals and humans [3–5]. Genetic or pharmacological inhibition of NF-κB pathways and cytokine neutralisation reverses insulin resistance [4, 6]. Previous studies demonstrated that inflammation-induced activation of protein kinases, such as IκB kinases (IKKs), c-jun N-terminal kinases (JNKs) and extracellular signal-regulated kinases (ERKs), inhibits tyrosine phosphorylation of IRS1 and suppresses downstream insulin signalling. This results in inefficient glucose uptake and use [7–9]. Thus, it is critical to identify the target(s) and mechanism(s) that initiate inflammatory responses leading to insulin resistance.
Insulin resistance is frequently associated with obesity [10] and dyslipidaemia [11]. Obesity is thought to be the most important identified factor contributing to insulin resistance [12]. Dyslipidaemia, characterised by abnormal amounts and composition of lipids and/or lipoproteins in blood, represents an additional risk factor for insulin resistance. NEFAs and triacylglycerols are elevated in obese and diabetic individuals and animals. Previous studies demonstrated that exposure to elevated NEFA causes activation of inflammatory signals and production of cytokines in multiple cell types, and these findings have been correlated with impairments of insulin actions [13, 14]. Thus, circulating lipid and associated inflammation appear to be a link between nutrient excess and insulin resistance. Recent studies have suggested that the induction of inflammatory pathways by dietary factors may mediate through the membrane receptors, including toll-like receptors (TLRs) [15].
TLRs play a central role in the innate immune response by recognising conserved patterns in diverse pathogenic molecules and activating pro-inflammatory signalling pathways [16, 17]. TLR2, a subclass of TLRs, is the main receptor recognising components of bacterial cell wall, lipoproteins and lipopeptides. Ligand-induced dimerisation of TLR2 with either TLR1 or TLR6 triggers recruitment of adaptor proteins, following a cascade of kinase activation, ultimately leading to activation of NF-κB and production of pro-inflammatory cytokines. The binding specificity of TLR2 with lipoprotein ligand is mediated through its dimerisation with either TLR1 or TLR6. The TLR2–TLR1 complex recognises triacylated lipoprotein, whereas the TLR2–TLR6 complex senses diacylated lipoprotein [18]. Thus, the number of acyl chain of lipoproteins is finely differentiated by TLR1–TLR2 and TLR2–TLR6. In addition to the number of acyl chains, other characteristics, such as length and ester bond of acyl chains, are critical for the biological activity of TLR2-dependent signalling [19, 20]. These results suggest that the fatty acid composition plays a central role in ligand recognition and receptor activation for TLR2. Furthermore, other studies demonstrated that TLR2 is involved in NEFA-induced insulin resistance [21–23]. It is reasonable to postulate that the fatty acid moiety from nutrients could potentially activate TLR2 and transduce the inflammatory signals.
Although the predominant site of TLR2 production is on cells of the innate immune system [17], TLR2 is found on a number of insulin-responsive cells, including adipose, skeletal muscle and liver cells [24, 25]. However, the roles of TLR2 in initiating and maintaining inflammatory-associated insulin resistance and energy homeostasis in vivo have not been established. Here, we show that mice with TLR2 deficiency are protected from developing insulin resistance and adiposity. The increased insulin sensitivity is associated with increased insulin-signal transduction, improved glucose metabolism, and reduced inflammatory cytokine expression and related signalling specifically in the liver.
Methods
Mice
Mice deficient in TLR2 (Tlr2 −/−) were kindly provided by S. Akira [26] and maintained on a C57BL/6 genetic background. Studies were carried out using both male and female Tlr2 −/− mice and age-matched wild-type (WT) C57BL/6 mice, obtained from National Cheng Kung University Laboratory Animal Center. Leptin-deficient (ob/ob) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were fed ad libitum either regular chow (RC) (Purina Laboratory Rodent Diet 5001, PMI Nutrition International, Richmond, IN, USA) or a high-fat (HF) diet (58Y1; TestDiet, Richmond, IN, USA).
Tissue collection and RNA analysis
Tissues were collected and stored in RNAlater (Ambion, Austin, TX, USA), and RNA was extracted using the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). Samples of mRNA were analysed with SYBR Green-based real-time quantitative RT-PCR (Applied Biosystems, Foster City, CA, USA), with β-actin or cyclophilin A as the reference gene in each reaction. Sequences of the primers used for RT-PCR assays are shown in Electronic supplementary material (ESM) Table 1.
Protein analysis
For the insulin signalling, 62.5 mU/kg insulin was administered through the portal vein, and muscle tissues were collected 5 min after injection as described by Hirosumi et al. [27]. Liver and WAT tissue samples were collected 2 min after injection with 200 and 500 mU/kg insulin through the portal vein, respectively. Further methods can be found in the ESM.
Results
Upregulation of TLR2 and inflammatory cytokines in WAT and liver of obese mice
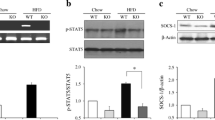
To examine the association of inflammation with obesity and insulin resistance, we determined the gene expression of inflammatory mediators in the major insulin-responsive tissues of ob/ob mice. We found that mRNA expression of Tnf was significantly elevated in WAT and liver, but not in muscle, of ob/ob mice, compared with those of WT controls (Fig. 1a). Expression of Il1b and Il6 mRNA was increased in WAT, but not in liver and muscle, of ob/ob mice. The upregulation of inflammatory genes was accompanied by significantly increased expression of TLR2 and moderate induction of TLR4 expression as shown in levels of both mRNA and protein (Fig. 1b,c). Consistent with this, the upregulation of TLR2 was evident in WAT and liver, but not in muscle. These results imply that obesity and nutrient excess elicit upregulation in expression of inflammatory cytokines and TLR2 predominantly in WAT and liver. This prompted us to test directly whether TLR2 and its induced inflammation are involved in metabolic disturbances.

Expression of inflammatory cytokines and TLRs in WAT, liver and gastrocnemius muscle of ob/ob mice. (a) Il1b, Il6 and Tnf, and (b) Tlr2 and Tlr4 mRNA levels in tissues from male ob/ob mice (n = 6–8) relative to samples from WT mice (n = 6–10). c Protein levels of TLR2 and TLR4 in tissues from male ob/ob and WT mice. Samples from representative animals are shown in the western blot, with each lane representing one animal. The intensities of the bands, quantified densitometrically relative to WT, are shown with sample number in parentheses. White bars, WT; grey bars, ob/ob. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with WT mice
Increased insulin sensitivity in Tlr2 −/− mice
To investigate the role of TLR2 in glucose homeostasis, we examined the plasma levels of glucose and insulin in Tlr2 −/− and WT mice. Plasma glucose and insulin levels after fasting were both significantly lower in Tlr2 −/− mice than in those of RC-fed WT mice (Fig. 2a,b). Consistent with these findings, Tlr2 −/− mice fed an HF diet had significantly lower glucose and insulin levels relative to their WT controls. To assess the effect of TLR2 deficiency on the whole-body glucose utilisation, we performed an OGTT. Tlr2 −/− mice cleared glucose faster than WT mice, regardless of diet, indicating improved glucose tolerance in Tlr2 −/− mice (Fig. 2c,d). This improved glucose tolerance was accompanied by significantly decreased plasma insulin levels during OGTT of Tlr2 −/− mice, regardless of diet (Fig. 2e,f). Thus, the insulin-resistance index calculated from the OGTT was significantly lower in Tlr2 −/− mice relative to their WT controls with either diet (Fig. 2g). Consistent with this, insulin tolerance tests (ITT) showed that glucose-lowering effects of insulin were improved in Tlr2 −/− mice regardless of diet (Fig. 2h,i). These results demonstrated that the absence of TLR2 increases glucose tolerance and insulin sensitivity regardless of feeding mice RC or an HF diet.

Glucose homeostasis and insulin sensitivity in Tlr2 −/− mice. a Fasting plasma glucose in male mice fed RC and HF diets (WT RC, n = 15; Tlr2 −/− RC, n = 16; WT HF, n = 16; Tlr2 −/− HF, n = 12). b Fasting insulin levels in male mice fed RC and HF diets (WT RC, n = 8; Tlr2 −/− RC, n = 7; WT HF, n = 14; Tlr2 −/− HF, n = 9). (c,d) Plasma glucose during OGTT in male mice fed (c) RC and (d) HF diets. (e,f) Insulin levels during OGTT in male mice fed (e) RC and (f) HF diets. g Insulin-resistance (IR) index during OGTT in male mice fed RC and HF diets. (h,i) ITT results for male mice fed (h) RC and (i) HF diets. White bars/symbols, WT; black bars/symbols, Tlr2 −/−. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with WT mice
Decreased body weight and fat mass in Tlr2 −/− mice
We next asked whether loss of TLR2 alters energy homeostasis in mice. The growth curve of Tlr2 −/− mice was the same as that of WT mice when mice were fed RC, but significantly differed in mice fed the HF diet (Fig. 3a,b). Body composition analysis by echoMRI identified significantly reduced fat mass and increased lean mass in aged (11 month old) Tlr2 −/− mice, while no difference in fat and lean mass was found between young (2 month old) Tlr2 −/− and WT mice (Fig. 3c). Furthermore, we found significant decreases in individual fat mass of aged Tlr2 −/− mice (Fig. 3d). The intra-abdominal, including gonadal and retroperitoneal, WAT masses in Tlr2 −/− mice were about 35% of those in WT mice. A similar reduction was also observed in the subcutaneous inguinal WAT mass of Tlr2 −/− mice, although the difference did not reach statistical significance. The interscapular brown adipose tissue (BAT) weight of Tlr2 −/− mice was about 70% that of WT mice. The weights of major organ, including kidney, liver, spleen and heart, were indistinguishable between genotypes. Gastrocnemius skeletal muscle weight of Tlr2 −/− mice was significantly increased. These data indicate that the absence of TLR2 causes reductions in fat deposition and content. While HF feeding increased body weight of WT mice, the increases in body weight of Tlr2 −/− mice were significantly diminished (Fig. 3b). Consistent with this, the individual WAT and BAT masses were significantly less in Tlr2 −/− mice fed an HF diet than in WT mice (Fig. 3e).

Body composition of Tlr2 −/− mice. (a,b) Body weights of female mice fed (a) RC (WT, n = 8; Tlr2 −/−, n = 7) and (b) HF diets (WT, n = 7; Tlr2 −/− , n = 7); at first time point after HF was started, p < 0.05; at time point 2, p < 0.01; at all subsequent time points, p < 0.001. c Body composition analyses of 2 and 11 month old male mice by MRI (2 month old mice, n = 5; WT 11 month old mice, n = 7; Tlr2 −/− 11 month old mice, n = 9). (d,e) Organ weights in (d) 11 month old female WT (n = 8) and Tlr2 −/− (n = 7) mice fed RC and (e) 7 month old female WT (n = 7) and Tlr2 −/− (n = 7) mice fed the HF diet. Gon, gonadal WAT; Ing, inguinal WAT; Ret, retroperitoneal WAT. Muscle indicates gastrocnemius skeletal muscle. (f,h) Morphology and (g,i) cell size distribution of gonadal WAT from WT and Tlr2 −/− mice fed (f,g) RC and (h,i) HF diets. The scale bar indicates 100 μm for all images. j In vitro adipogenesis in embryonic fibroblasts. Oil red O staining of plates after 8 days of differentiation. White bars/symbols, WT; black bars/symbols, Tlr2 −/−. *p < 0.05, **p < 0.01 and ***p < 0.001 compared with WT mice
Microscopically, adipocytes in gonadal WAT of RC-fed aged Tlr2 −/− mice were smaller than WT cells (mean area 821 μm2 in Tlr2 −/− vs 1503 μm2 in WT) (Fig. 3f,g). The numbers of adipocytes were similar in the Tlr2 −/− and WT mice (1.89 × 107 in TLR2−/− vs 1.73 × 107 in WT). HF feeding led to fat deposition in both Tlr2 −/− and WT mice and the difference in adipocyte size between genotypes was preserved (mean area 2242 μm2 in Tlr2 −/− vs 3276 μm2 in WT) (Fig. 3h,i). The decrease in adipocyte size of Tlr2 −/− mice under HF feeding was associated with a decrease in the number of adipocytes (1.21 × 107 in Tlr2 −/− vs 2.62 × 107 in WT). Thus, the deficiency of TLR2 brought a shift in the distribution of adipocyte sizes towards smaller adipocytes in WAT (Fig. 3g,i). The reduction in fat mass of Tlr2 −/− mice appears to result primarily from the reduction in adipocyte size with a modest decrease in adipocyte number. In vitro differentiation of embryonic fibroblasts showed that adipogenic ability judged by the formation of cells staining positive with Oil red O after hormone stimulation was preserved in Tlr2 −/− mice (Fig. 3j), ruling out the possibility of defective adipogenic programming with TLR2 deficiency.
The critical variables contributing to body weight maintenance include energy intake and expenditure. Metabolic analysis showed that Tlr2 −/− mice had similar daily food intake to WT mice (Fig. 4a), suggesting that Tlr2 −/− mice have normal energy absorption. Body temperature was modestly increased in male and significantly increased in female Tlr2 −/− mice, regardless of diet (Fig. 4b). Basal energy expenditure, as measured by oxygen consumption (\( \dot{V}{{\hbox{O}}_2} \)), during the dark phase was dramatically increased in Tlr2 −/− mice (Fig. 4c,d). As the increases in \( \dot{V}{{\hbox{O}}_2} \) and \( \dot{V}{\hbox{C}}{{\hbox{O}}_2} \) were coordinated, the respiratory exchange ratio was unchanged (Fig. 4e,f). Consistent with this, vertical rearing of both young and aged Tlr2 −/− mice during the dark phase was markedly increased (Fig. 4g). Ambulatory locomotor activity of aged Tlr2 −/− mice during both the dark and light phases was significantly increased (Fig. 4h). Thus, energy dissipation appeared increased in Tlr2 −/− mice.

Energy homeostasis in Tlr2 −/− mice. a Daily food intake in metabolism cages (n = 5 for all results). b Body temperature measured at 10:00 hours in WT and Tlr2 −/− mice (RC male [M] mice, n = 8; WT RC female [F] mice, n = 8; Tlr2 −/− F mice, n = 7; WT HF M mice, n = 8; Tlr2 −/− M mice, n = 5; WT HF F mice, n = 7; Tlr2 −/− F mice, n = 7. (c) Oxygen consumption (\( \dot{V}{{\hbox{O}}_2} \)) and (d) mean AUC; (e) respiratory exchange ratio (RER) and (f) mean 12 h values, of 2 month old male mice in indirect calorimetry over 48 h (WT, n = 5; Tlr2 −/−, n = 4). Bold bars on the x-axes represent the dark phases. (g, h) Locomotor activity measured as (g) the vertical rearing and (h) the ambulatory activity, across a 30 min period of observation (2 month old mice, n = 5; WT 11 month old mice, n = 7; Tlr2 −/− 11 month old mice, n = 9; D, dark; L, light). White bars/symbols, WT; black bars/symbols, Tlr2 −/−. *p < 0.05, **p < 0.01,***p < 0.001 and † p = 0.0695 compared with WT mice
Lipid profile and inflammatory mediators in Tlr2 −/− mice
WAT actively secretes signalling molecules, including NEFA, adipokines and inflammatory molecules, into the circulation and communicates with other organs to regulate insulin sensitivity [28]. Fasting plasma triacylglycerol and NEFA levels were not distinguishable between genotypes in mice fed RC, and were significantly lower in Tlr2 −/− mice than in WT mice fed an HF diet (Fig. 5a,b). Plasma cholesterol levels were not different between genotypes, regardless of diet (Fig. 5c). Circulating leptin and resistin levels were not distinguishable between genotypes in mice fed RC, but were reduced in Tlr2 −/− mice fed an HF diet (Fig. 5d,e). Adiponectin levels in circulation were comparable between Tlr2 −/− and WT mice, regardless of diet (Fig. 5f). While circulating inflammatory molecules, including IL-6, monocyte chemotactic protein-1 (MCP-1) and TNF-α, were higher in mice fed an HF diet than in those fed RC, no difference was detectable between genotypes, regardless of diet (Fig. 5g–i). Although the decreased levels of plasma triacylglycerol, NEFA and resistin may account for increased insulin sensitivity in Tlr2 −/− mice upon HF feeding, the increased insulin sensitivity was, however, observed in Tlr2 −/− mice fed RC despite any significant change in plasma triacylglycerol, NEFA and resistin. These results suggest that the deficiency of TLR2 has limited effects on systemic levels of inflammatory mediators.

Lipid profiles and inflammatory mediators in Tlr2 −/− mice. Plasma levels of (a) triacylglycerol, (b) NEFA, (c) cholesterol, (d) leptin, (e) resistin, (f) adiponectin, (g) IL-6, (h) MCP-1 and (i) TNF-α in male mice fed RC and HF diets (WT, n = 8; Tlr2 −/−, n = 5). White bars, WT; black bars, Tlr2 −/−. *p < 0.05 and **p < 0.01 for Tlr2 −/− mice compared with WT mice
Attenuated local inflammatory cytokine expression and signalling in Tlr2 −/− mice
Although the deficiency of TLR2 did not elicit apparent differences in the systemic levels of inflammatory molecules, insulin sensitivity can be affected through the change in local insulin-responsive tissues. We next examined whether the absence of TLR2 would attenuate local inflammation. Tlr2 −/− RC-fed mice exhibited, in liver, a significant reduction in expression of Il6 and a tendency towards decreased Il1b expression, but no change in expression of Tnf and macrophage marker Emr1 (Fig. 6a). No difference in expression of these cytokines was identified in WAT and muscle of Tlr2 −/− mice. Interestingly, dramatic reductions in expression of Il1b, Il6 and Emr1 as well as a tendency towards a decrease in expression of Tnf were observed in WAT and liver of Tlr2 −/− mice fed the HF diet (Fig. 6b). Expression of these genes, except Tnf, remained unaltered in muscle of Tlr2 −/− HF-fed mice (Fig. 6b). Because TLR2 has been demonstrated to transduce signal to activate protein kinases, including IKK, JNK and ERK [7–9], we examined their phosphorylation in the insulin-responsive tissues. No difference in phosphorylation of IKKα/β and JNK between WT and Tlr2 −/− mice was detectable in WAT, liver and muscle, regardless of diet (Fig. 6c,d). While phosphorylation of ERK1/2 was similar in WAT and muscle in the WT and Tlr2 −/− mice fed RC, its phosphorylation was markedly decreased in the liver of Tlr2 −/− mice (Fig. 6c). The decrease in hepatic ERK1/2 phosphorylation was not accompanied by the change of ERK1/2 protein content in the liver. Furthermore, the liver-specific decrease in ERK1/2 phosphorylation was also observed in Tlr2 −/− mice fed the HF diet (Fig. 6d). Thus, the absence of TLR2 attenuates basal inflammatory cytokine expression and signalling specifically in the liver.

Local inflammatory cytokine expression and signalling in Tlr2 −/− mice. Il1b, Il6, Tnf and Emr1 mRNA levels in Tlr2 −/− (n = 5–7) tissues relative to WT (n = 7–8) of mice fed (a) RC and (b) HF diets. White bars, WT; black bars, Tlr2 −/−. Immunoblot analyses on phosphorylation of IKKα/β, JNK and ERK1/2 and protein content of ERK1/2 in gonadal WAT, liver and gastrocnemius muscle from WT and Tlr2 −/− mice fed (c) RC and (d) HF diets. Immunoblot analyses on phosphorylation at Ser307 and Ser612 of IRS1 in the liver of WT and Tlr2 −/− mice on (e) RC and (f) HF diets. Each band represents a tissue extract from a single mouse. The intensities of the bands, quantified densitometrically relative to WT, are shown with the sample number in parentheses. *p < 0.05, **p < 0.01 and ***p < 0.001 for Tlr2 −/− mice compared with WT mice
Inflammation-induced activation of IKK, JNK and ERK has been shown to phosphorylate IRS1 at its serine residue(s), which further interrupts tyrosine phosphorylation on IRS1 and suppresses insulin signalling [7–9]. To further evaluate whether the attenuated inflammatory cytokine expression and signalling in the liver of Tlr2 −/− mice correlates with serine phosphorylation of IRS1, we detected the phosphorylation at Ser307 and Ser612 of IRS1. No difference in phosphorylation at Ser307 and Ser612 of IRS1 between WT and Tlr2 −/− livers was detected, regardless of diet (Fig. 6e,f). These results suggest that TLR2 deficiency did not affect phosphorylation at Ser307 or Ser612 of IRS1 in the liver.
Improved insulin signalling specifically in the liver of Tlr2 −/− mice
To determine which insulin-responsive tissue(s) exhibited improved insulin signalling in Tlr2 −/− mice, we examined the insulin action in mice fed RC. Insulin-stimulated phosphorylation at Ser473 of protein kinase B (Akt) was increased in liver, but not in WAT and muscle of Tlr2 −/− mice, compared with those of WT mice (Fig. 7a). This was accompanied by increased tyrosine phosphorylation of IRS1 and association of IRS1: phosphoinositide 3-kinase (PI3K) complex in the livers of Tlr2 −/− mice after insulin stimulation. However, the increases in IRS1 tyrosine phosphorylation and IRS1:PI3K association were not found in WAT and muscle of Tlr2 −/− mice. Consistently, the liver-specific increases in insulin-stimulated Akt phosphorylation, IRS1 tyrosine phosphorylation and IRS1:PI3K association (data not shown) were also observed in Tlr2 −/− mice fed HF diet (Fig. 7b). Thus, liver appears to be the major tissue with increased insulin signalling in Tlr2 −/− mice.

Insulin signalling in Tlr2 −/− mice. Immunoblot analyses on phosphorylation at Ser473 of Akt, and detection of IRS1 phosphotyrosine level and IRS1:PI3K association by IRS1 immunoprecipitation in mice fed (a) RC and (b) HF diets. Each band represents a tissue extract from a single mouse. The intensities of the bands relative to WT after insulin stimulation are shown with the sample number in parentheses. Ins, insulin. *p < 0.05 and **p < 0.01 for Tlr2 −/− mice compared with WT mice
Improved glucose metabolism in the liver of Tlr2 −/− mice
To determine the functional correlates of increased insulin signalling in the livers of Tlr2 −/− mice, we determined aspects of hepatic glucose metabolism. Pyruvate tolerance tests showed that glucose levels were significantly lower in Tlr2 −/− mice in response to the administration of pyruvate, a major gluconeogenesis substrate (Fig. 8a). We next assessed the effect of TLR2 deficiency in primary hepatocytes isolated from Tlr2 −/− and WT mice. TLR2 deficiency significantly reduced pyruvate-induced glucose release into the medium by primary hepatocytes (Fig. 8b). To examine the functional effects of TLR2 deficiency on hepatocyte insulin responsiveness, the ability of insulin to inhibit hepatocyte glucose production was determined. Glucose production by WT hepatocytes was reduced by ∼20% within this time period in response to insulin (Fig. 8c). The inhibition of glucose secretion by insulin in Tlr2 −/− hepatocytes was significantly increased by 50% in comparison with WT hepatocytes. Thus, increased insulin signalling secondary to TLR2 deficiency resulted in an enhancement of insulin to suppress hepatocyte glucose secretion. Together, these results indicate Tlr2 −/− mice have reduced hepatic glucose production and increased response to insulin.

Glucose metabolism and triacylglycerol content in liver and muscle of Tlr2 −/− mice. a Plasma glucose levels during PTT in male mice fed RC (n = 4 each). b Basal and pyruvate-induced glucose release into the medium after 4 h incubation in primary hepatocytes isolated from Tlr2 −/− and WT mice. c Insulin-suppressed glucose release into the medium after 16 h of incubation in primary hepatocytes isolated from Tlr2 −/− and WT mice. Results are expressed as the percentage inhibition of glucose secretion. d Basal and insulin-stimulated glucose uptake in isolated gastrocnemius muscle from Tlr2 −/− and WT mice. Results are derived from two independent experiments performed in triplicate. e Triacylglycerol (TG) content in liver and gastrocnemius muscle of WT (n = 8) and Tlr2 −/− (n = 10) mice. White bars/symbols, WT; black bars/symbols, Tlr2 −/−. *p < 0.05, **p < 0.01 and ***p < 0.001
To determine the insulin sensitivity in the skeletal muscle, we performed the in vitro glucose uptake assay. Basal glucose uptake in muscle in vitro was similar between Tlr2 −/− and WT mice (Fig. 8d). Insulin-stimulated glucose uptake in muscle of Tlr2 −/− mice was 1.6 times those of WT, although the difference did not reach the statistical significance. Consistent with this, triacylglycerol content in liver, which is associated with hepatic insulin resistance, was significantly reduced in Tlr2 −/− mice, while muscle triacylglycerol content was not altered in Tlr2 −/− mice (Fig. 8e).
Discussion
Substantial evidence indicates inflammation as a key mechanism linking metabolic disturbance to nutrient excess. To establish this link, we hypothesised that TLR2 transduces the inflammatory signals and further contributes to insulin resistance. Our findings recapitulated the observation that obesity is associated with elevated expression of inflammatory cytokines in WAT and liver. Moreover, mice lacking TLR2 exhibited enhanced insulin sensitivity, which is accompanied by increased transduction of insulin signal, improved glucose metabolism, and attenuated inflammatory cytokine expression and related signalling specifically in the liver. Thus, our work has identified TLR2 as a key mediator of hepatic inflammation-related signalling and insulin resistance.
Body weight represents the homeostasis between energy intake and expenditure. The deficiency of TLR2 caused a reduction in body weight and fat mass without alteration in food intake. Increased energy expenditure and locomotor activity, as well as body temperature, point to increased energy dissipation in Tlr2 −/− mice. It is known that increased physical activity can influence the insulin sensitivity of skeletal muscle, which is consistent with our findings of a modest increase of insulin-stimulated glucose uptake in Tlr2 −/− muscle. However, the differences in body weight and fat mass were not noticeable unless mice were aged up to 11 months or stimulated with a high-energy diet. The reductions in WAT mass and adipocyte size of Tlr2 −/− mice could result from the defect of pre-adipocytes differentiating into adipocytes and/or maturation of adipocytes. To address the bona fide role of TLR2 in adipocyte differentiation and maturation, we subjected embryonic fibroblasts to adipogenesis in vitro. TLR2 deficiency did not influence the abilities of adipocyte differentiation and triacylglycerol accumulation, suggesting that these properties are not affected by TLR2 deficiency. Although the mechanism involved in increased energy expenditure in Tlr2 −/− mice is not clear, the reduced deposition of excess energy in the body potentially contributes to better metabolic profiles and glucose metabolism.
Improvement of insulin sensitivity with TLR2 deficiency is present in mice fed RC, suggesting TLR2-induced insulin resistance takes place in the basal physiological condition without the need of further HF stimulation. This raises the speculation that TLR2 ligand and the signalling it mediates are present in the basal state. Although the actual endogenous TLR2 ligands have not been identified conclusively, it has been demonstrated that TLR2 recognises a large number of lipid-containing molecules [18], as well as endogenous proteins released from damaged tissues or injured cells [29]. It is interesting to note that the critical portion of many TLR2 ligands contains a similar fatty acid component. Removal or change of these fatty acids results in loss of TLR2 activation capability [30], implicating an important role of these fatty acids in ligand recognition and receptor activation. Particularly, palmitic and stearic acids are known to be major fatty acids acylated in the lipoprotein or lipopeptide to activate TLR2 [31, 32]. NEFA possess the capacity to induce stress/inflammatory signals not only in immune cells but also other cell types and tissues [13, 14, 33]. This phenomenon is of particular importance in conditions of nutrient excess such as obesity and diabetes or after ingestion of a fatty meal.
Inflammation-induced activation of protein kinases, such as IKK, JNK and ERK, has been shown to inhibit tyrosine phosphorylation of IRS1 [7–9] and suppress insulin action and its downstream signalling. Serine phosphorylation of IRS1 is a general mechanism to interrupt IRS1 function and insulin-signal transduction [34, 35]. For example, phosphorylation of IRS1 at Ser307 reduces IRS1 coupling to activated insulin receptors [8] and enhances IRS1 degradation [36]. Phosphorylation of IRS1 at Ser612 decreased PI3K docking to IRS1 [37]. IRS1 Ser307 can be phosphorylated by IKK and JNK [7, 8], whereas IRS1 Ser612 can be phosphorylated by ERK [9]. Our results showing enhanced insulin-stimulated Akt Ser473 phosphorylation and IRS1 tyrosine phosphorylation specifically in the liver of Tlr2 −/− mice provide a biochemical correlate for increased hepatic insulin sensitivity. In the search for protein kinases involved in insulin signalling that are induced by inflammatory mediators, we did not detect alterations in the activation of JNK and IKK in the livers of Tlr2 −/− mice. ERK activation was dramatically reduced in the livers of Tlr2 −/− mice, providing a possible link between TLR2 and hepatic inflammation. However, further investigation of the hypothesised phosphorylation target on IRS1 did not reveal any change in phosphorylation of Ser307 and Ser612. These results suggest that the hypothesised phosphorylation target on IRS1 mediated through TLR2 is not Ser307 or Ser612. Nevertheless, our results implicate ERK as a downstream intracellular mediator of the TLR2-induced inflammatory response influencing the insulin signal pathway through IRS1 modification.
Our observation of improved insulin sensitivity in the absence of TLR2 supports and elaborates previous findings. For example, palmitate induced IL-6 production and inflammatory signalling, leading to inhibition of insulin-activated signal transduction through TLR2 in myotubes [21]. In addition, inhibition of TLR2 expression by TLR2 antisense oligonucleotide improves insulin sensitivity and signalling in muscle and WAT from mice fed the HF diet [38]. However, tissue specificity and transient treatment of antisense oligonucleotide limit the interpretation of long-term effect of TLR2 on complex physiological homeostasis. Although mice lacking TLR2 have recently been found to be protected from diet-induced adiposity and systemic insulin resistance [39], mechanistic insight and tissue specificity for the improvement of insulin sensitivity with TLR2 deficiency have been lacking. Collectively, these findings implicate TLR2 as a key mediator of inflammation, causing insulin resistance under conditions of nutrient excess in many key insulin-responsive tissues.
Despite the apparent upregulation of inflammatory cytokines in WAT of ob/ob mice, the induction of inflammatory cytokines and TLR2 was also present in the livers of ob/ob mice in our study. These findings suggest that the nutrient-induced inflammatory response and insulin resistance takes place in both WAT and liver. In support of this hypothesis, TLR2-deficiency-associated decreases in inflammatory cytokine expression and signalling and enhancement of insulin signalling were conspicuous in the liver. In contrast, the induction of inflammatory cytokines was not evident in the muscle of ob/ob mice. Consistent with this, the gene expression and signalling molecules related to inflammation and insulin resistance in muscle appeared unchanged in Tlr2 −/− mice. Thus, the modification of peripheral insulin sensitivity through TLR2-induced inflammatory cytokine expression and signalling is likely to be mediated primarily in the liver.
Recently, the role of TLR4 in the cross-talk between nutrient-induced inflammation and insulin resistance has been investigated [40–43], and the hypothesised location for TLR4 action mediating the impairment of insulin sensitivity is predominantly in WAT. Different TLR production profiles affect the responsiveness of particular cell types and tissues to the ligand stimulation. In addition, different TLR ligands may induce activation of different downstream signalling pathways, leading to a diverse array of target gene expression and cellular responses through distinct TLRs. Given the differences in ligand preference, signalling pathways and tissue distribution between TLR2 and TLR4, it is reasonable to speculate that TLR2 and TLR4 may receive different ligand stimulation from nutrient factors and transduce inflammatory signals. Furthermore, there was no compensatory alteration in the gene expression of Tlr4 in WAT, liver and muscle (data not shown). Our results unequivocally provide TLR2 as another TLR family receptor involved in the interface of inflammatory response and metabolic disturbance.
In conclusion, our results demonstrated that TLR2 contributes to upregulation of inflammatory cytokine expression and signalling and interference of insulin action specifically in the liver. Although the detailed mechanism by which factors activate TLR2 or interact with its associated factors is not known, the results presented here represent a new paradigm that endogenous molecule(s) can utilise the innate immune receptor TLR2 to trigger the local inflammation-related signalling in the liver and subsequently affect systemic metabolism. Our findings provide a rationale for the development of the hepatic inflammatory response and insulin resistance in response to nutrient factors.
Abbreviations
- Akt:
-
Protein kinase B
- BAT:
-
Brown adipose tissue
- ERK:
-
Extracellular signal-regulated kinase
- HF:
-
High fat
- IKK:
-
IκB kinase
- ITT:
-
Insulin tolerance test
- JNK:
-
c-Jun N-terminal kinase
- MCP-1:
-
Monocyte chemotactic protein-1
- NF-κB:
-
Nuclear factor kappa B
- PI3K:
-
Phosphoinositide 3-kinase
- RC:
-
Regular chow
- TLR:
-
Toll-like receptor
- WAT:
-
White adipose tissue
References
Wellen KE, Hotamisligil GS (2005) Inflammation, stress, and diabetes. J Clin Invest 115:1111–1119
Shoelson SE, Lee J, Goldfine AB (2006) Inflammation and insulin resistance. J Clin Invest 116:1793–1801
Kim JK, Kim YJ, Fillmore JJ et al (2001) Prevention of fat-induced insulin resistance by salicylate. J Clin Invest 108:437–446
Cai D, Yuan M, Frantz DF et al (2005) Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 11:183–190
Shoelson SE, Lee J, Yuan M (2003) Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord 27(Suppl 3):S49–S52
Yuan M, Konstantopoulos N, Lee J et al (2001) Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkβ. Science 293:1673–1677
Gao Z, Hwang D, Bataille F et al (2002) Serine phosphorylation of insulin receptor substrate 1 by inhibitor kappa B kinase complex. J Biol Chem 277:48115–48121
Aguirre V, Uchida T, Yenush L, Davis R, White MF (2000) The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275:9047–9054
De Fea K, Roth RA (1997) Modulation of insulin receptor substrate-1 tyrosine phosphorylation and function by mitogen-activated protein kinase. J Biol Chem 272:31400–31406
Stamler R, Stamler J, Riedlinger WF, Algera G, Roberts RH (1978) Weight and blood pressure. Findings in hypertension screening of 1 million Americans. JAMA 240:1607–1610
Bonora E, Kiechl S, Willeit J et al (1998) Prevalence of insulin resistance in metabolic disorders: the Bruneck Study. Diabetes 47:1643–1649
Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106:473–481
Ghanim H, Aljada A, Hofmeyer D, Syed T, Mohanty P, Dandona P (2004) Circulating mononuclear cells in the obese are in a proinflammatory state. Circulation 110:1564–1571
Jove M, Planavila A, Laguna JC, Vazquez-Carrera M (2005) Palmitate-induced interleukin 6 production is mediated by protein kinase C and nuclear-factor kappaB activation and leads to glucose transporter 4 down-regulation in skeletal muscle cells. Endocrinology 146:3087–3095
Tschop M, Thomas G (2006) Fat fuels insulin resistance through Toll-like receptors. Nat Med 12:1359–1361
Akira S, Takeda K, Kaisho T (2001) Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol 2:675–680
Medzhitov R (2001) Toll-like receptors and innate immunity. Nat Rev Immunol 1:135–145
Akira S (2003) Mammalian toll-like receptors. Curr Opin Immunol 15:5–11
Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Ulmer AJ (2005) Lipopeptide structure determines TLR2 dependent cell activation level. FEBS J 272:6354–6364
Buwitt-Beckmann U, Heine H, Wiesmuller KH et al (2006) TLR1- and TLR6-independent recognition of bacterial lipopeptides. J Biol Chem 281:9049–9057
Senn JJ (2006) Toll-like receptor-2 is essential for the development of palmitate-induced insulin resistance in myotubes. J Biol Chem 281:26865–26875
Murakami K, Bujo H, Unoki H, Saito Y (2007) High fat intake induces a population of adipocytes to co-express TLR2 and TNFalpha in mice with insulin resistance. Biochem Biophys Res Commun 354:727–734
Nguyen MT, Favelyukis S, Nguyen AK et al (2007) A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 282:35279–35292
Lin Y, Lee H, Berg AH, Lisanti MP, Shapiro L, Scherer PE (2000) The lipopolysaccharide-activated toll-like receptor (TLR)-4 induces synthesis of the closely related receptor TLR-2 in adipocytes. J Biol Chem 275:24255–24263
Lang CH, Silvis C, Deshpande N, Nystrom G, Frost RA (2003) Endotoxin stimulates in vivo expression of inflammatory cytokines tumor necrosis factor alpha, interleukin-1beta, -6, and high-mobility-group protein-1 in skeletal muscle. Shock 19:538–546
Takeuchi O, Hoshino K, Kawai T et al (1999) Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity 11:443–451
Hirosumi J, Tuncman G, Chang L et al (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Rosen ED, Spiegelman BM (2006) Adipocytes as regulators of energy balance and glucose homeostasis. Nature 444:847–853
Rubartelli A, Lotze MT (2007) Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28:429–436
Brightbill HD, Libraty DH, Krutzik SR et al (1999) Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 285:732–736
Fujita M, Into T, Yasuda M et al (2003) Involvement of leucine residues at positions 107, 112, and 115 in a leucine-rich repeat motif of human Toll-like receptor 2 in the recognition of diacylated lipoproteins and lipopeptides and Staphylococcus aureus peptidoglycans. J Immunol 171:3675–3683
Muhlradt PF, Kiess M, Meyer H, Sussmuth R, Jung G (1997) Isolation, structure elucidation, and synthesis of a macrophage stimulatory lipopeptide from Mycoplasma fermentans acting at picomolar concentration. J Exp Med 185:1951–1958
Nguyen MT, Satoh H, Favelyukis S et al (2005) JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3 T3-L1 adipocytes. J Biol Chem 280:35361–35371
Gual P, Le Marchand-Brustel Y, Tanti JF (2005) Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 87:99–109
Taniguchi CM, Emanuelli B, Kahn CR (2006) Critical nodes in signalling pathways: insights into insulin action. Nat Rev Mol Cell Biol 7:85–96
Greene MW, Sakaue H, Wang L, Alessi DR, Roth RA (2003) Modulation of insulin-stimulated degradation of human insulin receptor substrate-1 by Serine 312 phosphorylation. J Biol Chem 278:8199–8211
Mothe I, Van Obberghen E (1996) Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J Biol Chem 271:11222–11227
Caricilli AM, Nascimento PH, Pauli JR et al (2008) Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J Endocrinol 199:399–406
Himes RW, Smith CW (2010) Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J 24:731–739
Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116:3015–3025
Tsukumo DM, Carvalho-Filho MA, Carvalheira JB et al (2007) Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56:1986–1998
Poggi M, Bastelica D, Gual P et al (2007) C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia 50:1267–1276
Kim F, Pham M, Luttrell I et al (2007) Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res 100:1589–1596
Acknowledgements
We thank A. Pendse, N. Takahashi and C.-H. Lee for discussions; Z.-H. Lin, H.-F. Jheng, the National Laboratory Animal Centre Pathology Core (C.-T. Liang), H.-T. Wu, Y.-H. Lee and the Taiwan Mouse Clinic for technical assistance; and S. Akira for kindly providing Tlr2 −/− mice. This work was supported by the grant from the National Health Research Institute (NHRI-EX98-9823SC).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Sequences of primers used for real-time quantitative RT-PCR (PDF 94 kb)
ESM 1
(PDF 26 kb)
Rights and permissions
About this article
Cite this article
Kuo, LH., Tsai, PJ., Jiang, MJ. et al. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia 54, 168–179 (2011). https://doi.org/10.1007/s00125-010-1931-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1931-5