
Abstract
Aims/hypothesis
The regulation of cortisol metabolism in vivo is not well understood. We evaluated the relationship between cortisol metabolism and insulin sensitivity, adjusting for total and regional fat content and for non-alcoholic fatty liver disease.
Materials and methods
Twenty-nine middle-aged healthy men with a wide range of BMI were recruited. We measured fat content by dual-energy X-ray absorptiometry and magnetic resonance imaging (MRI), liver fat by ultrasound and MRI, the hypothalamic-pituitary-adrenal axis by adrenal response to ACTH1-24, unconjugated urinary cortisol excretion, corticosteroid-binding globulin, and cortisol clearance by MS. We assessed insulin sensitivity by hyperinsulinaemic-euglycaemic clamp and by OGTT.
Results
Cortisol clearance was strongly inversely correlated with insulin sensitivity (M value) (r = −0.61, p = 0.002). Cortisol clearance was increased in people with fatty liver compared with those without (mean±SD: 243 ± 10 vs 158 ± 36 ml/min; p = 0.014). Multiple regression modelling showed that the relationship between cortisol clearance and insulin sensitivity was independent of body fat. The relationship between fatty liver and insulin sensitivity was significantly influenced by body fat and cortisol clearance.
Conclusions/interpretation
Cortisol clearance is strongly associated with insulin sensitivity, independently of the amount of body fat. The relationship between fatty liver and insulin sensitivity is mediated in part by both fatness and cortisol clearance.
Similar content being viewed by others

Introduction
Although many obese individuals resemble patients with excess cortisol exposure, plasma cortisol levels in overweight and obese subjects have been found to be either similar to or lower than those in non-obese individuals, suggesting that cortisol metabolism may be altered in some overweight or obese individuals [1–3]. Indeed, there are differences in cortisol metabolism between obese men and women [4] according to patterns of regional body fat distribution [5]. In obesity, inactivation of cortisol by 5α-reductase is enhanced, and this effect is offset by impaired metabolism of cortisol by 5β-reductase in women and enhanced conversion of cortisone to cortisol by 11β-reductase in men [4]. Patients with increased intra-abdominal fat have increased cortisol clearance, and cortisol clearance is correlated with intra-abdominal fat mass [5]. Several studies show that indices of hypothalamic–pituitary–adrenal (HPA) axis activity, including raised fasting cortisol concentrations and an increased adrenal response to ACTH1–24, are associated with insulin resistance independently of the effects of obesity [6, 7], suggesting that patterns of cortisol metabolism vary according to insulin sensitivity. Since overweight and obesity potentially confound the relationship between cortisol metabolism and insulin sensitivity, studies of the relationship between cortisol metabolism and insulin resistance need to adjust for total and regional fat content.
It is well recognised that obesity is associated with non-alcoholic fatty liver disease (NAFLD) and obese subjects with NAFLD are markedly insulin resistant [8–10], although the explanation for the marked insulin resistance with NAFLD is poorly understood [11]. Specifically, it is uncertain whether abnormal cortisol regulation or metabolism is responsible for the marked insulin resistance. To date, abnormalities of cortisol regulation have been described in NAFLD, including chronic activation of the HPA axis [12–15], increased urinary excretion of cortisol, and impaired suppression of cortisol [14]. However, whether these abnormalities occur because of insulin resistance, liver fat per se, or because of the association of increased body fat with NAFLD is uncertain.
Since there is evidence of increased clearance of cortisol with obesity [4], specifically with increased abdominal fat [5], we hypothesised that there would be increased cortisol clearance with insulin resistance. However, since NAFLD commonly occurs with obesity and insulin resistance, it is currently uncertain whether altered cortisol clearance occurring with abdominal obesity is mediated by the presence of NAFLD. Therefore, our aims were: (1) to examine the relationship between cortisol metabolism and insulin sensitivity; and (2) to investigate the effects of fatty liver and total and regional fat content on the relationship between cortisol metabolism and insulin sensitivity. We assessed cortisol metabolism using the adrenal response to ACTH1–24, unconjugated urinary cortisol excretion, and the plasma clearance of cortisol, together with insulin sensitivity, regional body fat and liver fat.
Subjects and methods
Recruitment and exclusion criteria
Twenty-nine healthy white middle-aged northern European male volunteers aged 42–64 years with a wide range of BMI were recruited from the general population. Study participants provided informed consent and the study was approved by the local research ethics committee. For additional information on subject recruitment (see the Electronic supplementary material [ESM]).
Body composition and metabolic syndrome features
Fasting plasma glucose was measured and a full lipid profile was established before an OGTT (see ESM). Measurements of glucose, lipid profile, insulin and NEFA were undertaken. Adiponectin, TNF-α, IL-6 and IL-1β were measured using ELISAs from R&D Systems (Minneapolis, MN, USA). The mean intra- and inter-assay CVs for the ELISAs were <3% in our hands. BMI was determined and waist measurement was measured midway between the costal margin and the iliac crest in expiration. Dual-energy X-ray absorptiometry (DEXA) scanning was undertaken on a Delphi W instrument (Hologic, Bedford, MA, USA) using a standard visual method to divide images into trunk, limbs and head. Total fat mass, truncal fat mass and lean mass were estimated from the DEXA scan measurements. A subgroup of 19 men underwent abdominal MRI to validate DEXA truncal fat measurements and to measure visceral and subcutaneous fat quantitatively. Images of five horizontal slices, each 5 cm apart, were taken through the abdomen, from the lower sternum to the pubic symphysis. Blood pressure was measured using a Marquette Dash 3000 monitor (GE Healthcare, Slough, Berks, UK).
Liver fat assessment
Liver ultrasonography was undertaken by one of two experienced radiologists. Scoring of the degree of hepatic steatosis for all scans was undertaken independently by both radiologists, by visual assessment of the degree of echogeneity. Scans were graded for the presence or absence of fatty liver. A subgroup of 16 individuals underwent quantitative assessment of liver fat by MRI (see ESM).
Measurement of insulin sensitivity in individual tissues (see ESM)
Muscle and liver
Muscle insulin sensitivity was measured as glucose infusion during the steady state of a hyperinsulinaemic–euglycaemic clamp. 2H-labelled glucose infusion (\({\left[ {{\text{6,\kern-1.6pt{6} - }}^{{\text{2}}} {\text{H}}_{{\text{2}}} } \right]}\)glucose; 170 mg, 1.7 mg/min; Cambridge Isotopes, Cambridge, MA, USA) was infused at the same time to determine hepatic insulin sensitivity, measured by insulin-mediated suppression of endogenous glucose production [16, 17]. The M value of insulin sensitivity was defined as the glucose infusion rate during the final 30 min of the test in mg kg−1 min−1. The isotopic enrichment of glucose was measured by GC-MS and total glucose production (Ra) and total glucose uptake (Rd) were calculated. Tracer to tracee ratios (TTR) were calculated as the ratios of the two areas from the MS data. TTR and plasma glucose values were smoothed using the method of optical segments [18]. For the Steele equations [19] 0.22 l/kg was used as the distribution volume of glucose to calculate Ra and Rd [20]. Glucose disappearance (Rd) during the steady state was used as the measure of insulin sensitivity in muscle. These measures were corrected for total body weight. Hepatic insulin sensitivity was measured as insulin-mediated suppression of glucose output in the early part of the clamp and was expressed as percentage suppression 60 min after commencing the insulin infusion; suppression was linear to this time point. The intra- and inter-assay CVs for the Delphia insulin assay ranged from 4 to 6% according to low, medium and high standards.
Fat
NEFA were measured fasting and during the OGTT to allow measurement of insulin-mediated suppression of NEFA concentration. Insulin sensitivity indices for NEFA were estimated using data from fasting and 1 and 2 h samples for insulin and NEFA concentrations, according to the established method proposed for calculating insulin sensitivity from data obtained during an OGTT [21].
HPA axis studies, measurement of unconjugated urinary cortisol and estimation of glomerular filtration rate (see ESM)
Fasting cortisol (09.00 hours) was measured using an in-house RIA in blood treated with lithium heparin [22]. An ACTH1–24 analogue, tetracosactrin (Synacthen; Alliance, Chippenham, Wilts, UK), was administered to determine the sensitivity of the HPA axis. A physiological dose of 1 μg of freshly prepared ACTH1–24 was used for the test [6]. A sample of urine was taken at this time in order to minimise circadian variation in cortisol metabolites and to estimate 24 h unconjugated urinary cortisol excretion [23]. The 24 h unconjugated urinary cortisol excretion was estimated as unconjugated urinary cortisol/urinary creatinine ×10 in a sample of urine provided at 09.00 h on the day of 2H-labelled cortisol administration. GFR was estimated by calculation using the Modification of Diet in Renal Disease Study Group equation [24].
Cortisol clearance
(see ESM) Cortisol clearance was measured by injecting a tracer bolus of \({\left[ {{\text{9,\kern-1.8pt{12},\kern-1.8pt{12} - }}^{{\text{2}}} {\text{H}}_{{\text{3}}} } \right]}\) cortisol (Cambridge Isotopes) [25]. The lipid-extracted samples were measured by liquid chromatography tandem mass spectrometry (LC/MS/MS) using a symmetrical C18 3.5 μm 2.1 × 50 mm column (Waters, Milford, MA, USA). Each measurement was repeated ten times to decrease errors. Eluted sterols were quantified on Quatro Ultima triple quadrupole mass spectrometer (Micromass, Manchester, UK) by multiple reaction monitoring (MRM) scans. A software program (Mass-Lynx, Waters) was used to integrate areas for each eluted sterol. Results were manually checked and the mean of six replicate measurements was used for each data point. Clearance of cortisol was calculated from the cortisol measurements and spiked sample measurements using a two-pool method as described by Tait [26].
Corticosteroid-binding globulin
(see ESM) Corticosteroid-binding globulin (CBG) was measured using a commercial RIA kit (code CBG-RIA-100; Biosource, Nivelles, Belgium).
Statistical analyses
All statistical analyses were performed using SPSS for Windows version 14.0 (SPSS Inc., Chicago, IL, USA). Student’s t tests were undertaken to compare mean values of normally distributed data. Where variables were not normally distributed, log transformation was undertaken to normalise the distribution. Multivariate linear regression modelling was undertaken to describe the measures that were independently associated with M value as the dependent (outcome) variable (see ESM). To avoid collinearity and to validate the model, the cortisol clearance data were transformed by the simple mathematical manipulation of centring [27] (see ESM).
Results
Baseline characteristics of the men are shown in Table 1 (see also ESM). Cortisol clearance was inversely related to the CBG concentration (r = −0.490, p = 0.018). Cortisol clearance was not associated with 09.00 h cortisol concentration (r = −0.10, p = 0.65), cortisol concentrations following the ACTH1–24 stimulation test (cortisol 0 min, r = −0.17, p = 0.43; cortisol 30 min, r = −0.24, p = 0.28; cortisol 60 min, r = −0.03, p = 0.91), unconjugated urinary cortisol excretion (r = −0.24, p = 0.36) or estimated GFR (r = −0.15, p = 0.49).
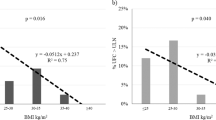
Steady-state insulin concentrations were achieved for all individuals during the last 30 min of the clamp when the M value (whole-body insulin sensitivity) was estimated. There was little variation in insulin concentration between individuals during the steady-state period (mean±SEM: 168 ± 9.5 pmol/l). There was a strong inverse correlation between cortisol clearance and M value (r = −0.61, p = 0.002) (Fig. 1). Figure 1 also shows cortisol clearance and M values in subjects with fatty liver and no fatty liver. There were also strong inverse relationships between cortisol clearance and insulin sensitivity in muscle (r = −0.53, p = 0.009) and fat (r = −0.42, p = 0.045). Cortisol clearance (mean±SD) was 270 ± 100 ml/min in the most insulin-resistant tertile and 150 ± 30 ml/min in the most insulin-sensitive tertile of whole-body glucose disposal (M value) (p = 0.004). There were no significant associations between the fasting, total or free cortisol concentrations, or the cortisol response to ACTH1–24, and individual components of the metabolic syndrome (blood pressure, fasting glucose, plasma triacylglycerol or HDL-cholesterol concentrations and waist circumference).

Relationship between insulin sensitivity and cortisol clearance in men with fatty liver (open circles) and normal subjects (closed circles)
CBG correlated with M value (r = 0.525, p = 0.010). Similar relationships were observed between CBG and tissue insulin sensitivity in muscle, fat and liver (muscle [Rd], r = 0.477, p = 0.021; fat, r = 0.424, p = 0.044; liver, r = 0.460, p = 0.027).
CBG concentrations were increased in subjects who were in the most insulin-sensitive tertile compared with the least sensitive tertile (50.5 ± 1.8 vs 40.3 ± 3.1 mg/l; p = 0.01).
Since subjects with fatty liver are insulin-resistant and often overweight or obese, we explored the relationship between fatty liver and measures of cortisol metabolism. Fourteen subjects had no evidence of fatty liver and 15 subjects had fatty liver. Liver ultrasound scores by the two radiologists, independently scoring the scans, showed that agreement was 96% (κ statistic 0.93, SE 0.14; p < 0.0001). A subgroup of 19 men underwent MRI scanning to validate the semiquantitative assessment of liver fat by ultrasound. These data (not shown) verified that we had established two groups by ultrasonography: no fatty liver and fatty liver. In comparison with the men without fatty liver, those with fatty liver were of a similar age, but had significantly higher BMI, higher blood pressure and poorer glucose tolerance (data not shown). The men with fatty liver were markedly and significantly more insulin-resistant in muscle, fat and liver than those without fatty liver (p < 0.001, p < 0.001 and p < 0.04, respectively). There was no difference in reported alcohol intake between the two groups. Fatty liver was associated with increased cortisol clearance (mean ± SD: fatty liver, 243 ± 104 ml/min; no fatty liver, 158 ± 36 ml/min; p = 0.02). There were no significant differences between groups in CBG concentration, 09.00 h cortisol concentration and 0, 30, 60 min cortisol concentration after ACTH1–24. Free cortisol concentrations, estimated by the ratio of total cortisol to CBG concentration, and the response to ACTH1–24, measured by both the peak cortisol concentration and the incremental increase in cortisol concentration, were similar in the two groups. For the subgroup of subjects who underwent abdominal MRI scanning, liver fat was quantitated and treated as a continuous variable. In support of the semiquantitative ultrasonographic data, liver fat by MRI measurement was correlated with M value (r = −0.53, p = 0.04), insulin sensitivity in fat (r = −0.52, p = 0.04) and insulin sensitivity in liver (r = −0.51, p = 0.05). Liver fat was not correlated with cortisol clearance (r = 0.25, p = 0.46) or CBG concentration (r = −0.27, p = 0.43).
Since there was a strong inverse univariate association between cortisol clearance and whole-body insulin sensitivity (Fig. 1), we studied the relationships between cortisol clearance and insulin sensitivity in multivariate models (see ESM). To determine the effects of total body fat, regional body fat and fatty liver on the relationship between cortisol clearance and insulin sensitivity (M value), multiple regression modelling was undertaken with the M value as the dependent variable. Age, CBG, body fat, fatty liver and cortisol clearance were included in the model as explanatory variables (Table 2). Variation in total fat and cortisol clearance explained 82% (R 2 = 0.82) of the variance in M value (model B in Table 2), whereas variation in truncal fat and cortisol clearance explained 85% (R 2 = 0.85) of the variance in M value (model A in Table 2). The regression model was repeated using stepwise linear regression to examine the relative contributions of cortisol clearance and body fat to the variance in insulin sensitivity. These data (not shown) confirmed that cortisol clearance explained an additional 16% of the variance in insulin sensitivity (over and above the contribution from the measure of body fat).
Thus, the regression modelling analyses showed that body fat and cortisol clearance were both independently associated with insulin sensitivity and that, with inclusion of body fat and cortisol clearance as explanatory variables, fatty liver was not independently associated with insulin sensitivity (Table 2). To verify this finding, we also included the quantitative MRI liver fat measurements in a regression model (from the subgroup of subjects who underwent abdominal MRI). This analysis confirmed and supported the liver fat ultrasonography-derived data. In a regression model that included total body fat, cortisol clearance and liver fat as explanatory variables and M value as the outcome variable, MRI liver fat was not associated with M value (B = −0.001, p = 0.15), whereas cortisol clearance was associated with M value (B = −4.38, 95% CI −8.67, −0.89, p < 0.05).
To explore further the relationship between fatty liver and insulin sensitivity, the regression analyses were repeated. We omitted, in turn and singly, first cortisol clearance and then the measure of body fat. These results showed that: (1) with cortisol clearance omitted, fatty liver became independently associated with M value (B = −0.13, 95% CI −0.25, −0.01, p = 0.035); and (2) when cortisol clearance was reinserted and a measure of body fat removed (truncal or total, depending upon which measure was originally in the model), fatty liver again became independently associated with M value (B = −0.30, 95% CI −0.43, −0.17, p < 0.001 for the measure of truncal fat removed). Thus, these two regression models suggested that the relationship between fatty liver and insulin sensitivity was influenced via a contribution from both fatness and cortisol clearance.
To verify the regression models shown in Table 2, the M value was also estimated according to lean body mass and the steady-state insulin concentration achieved during the clamp (adjusted M value = mg glucose disposal kg lean mass−1 min−1 divided by steady-state insulin concentration). (This adjusted M value variable was calculated because lean mass includes skeletal muscle mass and skeletal muscle is the principal tissue for glucose uptake during the clamp.) The results shown in Table 3 were very similar to those obtained with the more classical estimation of M value (Table 2).
Discussion
We have shown that increased cortisol clearance is strongly associated with decreased insulin sensitivity independently of the amounts of total or truncal body fat. To our knowledge, this study is the first to show that increased cortisol clearance is strongly associated with decreased insulin sensitivity, independently of body fat and fatty liver. Our multivariate analyses show that the relationship between fatty liver and insulin sensitivity is mediated in part both by body fat and by cortisol clearance.
There was a strong correlation between DEXA trunk fat and abdominal fat measurement by MRI (r = 0.6, p = 0.007) (see ESM). Other data also suggest that DEXA measurement of truncal fat is an excellent proxy measure for visceral fat quantity as estimated by computed tomography [28], and the use of DEXA enables investigators to obtain data on total body fat and lean mass. In our study, controlling for truncal fat did not alter the relationship between cortisol clearance and insulin resistance. Our data support the findings of a study showing that visceral adipose fat mass was not associated with indices of cortisol metabolism after adjusting for the effects of whole-body and liver fat [14]. Moreover, a study of portal vein and peripheral vein cortisol concentrations obtained from obese subjects undergoing bariatric surgery showed that cortisol concentrations were not increased in the portal circulation [29]. This suggests that visceral fat does not generate markedly greater amounts of cortisol than subcutaneous adipocytes in obesity and does not contribute markedly to cortisol clearance. Taken together with our findings, these data suggest that other tissues besides body fat may contribute to increased cortisol clearance with insulin resistance.
In obese subjects, liver ultrasound has 88% sensitivity and 94% specificity to detect fatty liver compared with histology [30], and many of our subjects were obese. Moreover, the kappa statistic showed excellent agreement between the two radiologists who independently scored the ultrasound scans (see ESM). Since we showed a marked increase in cortisol clearance with fatty liver and individuals with fatty liver are markedly insulin-resistant [10], we investigated the effect of including fatty liver in the regression models. The regression analyses (Table 2) show that fatty liver did not appear to be independently associated with insulin sensitivity when cortisol clearance and a measure of body fatness were included in the model. The regression analyses were repeated after omitting in turn cortisol clearance and the measure of body fat. The results from these analyses showed that fatty liver became independently associated with insulin sensitivity in both analyses. Thus, these data suggest that the relationship between fatty liver and insulin sensitivity is mediated both by cortisol clearance and by body fatness. Since we showed no strong associations between measures of insulin sensitivity and 09.00 h cortisol levels, ACTH-stimulated cortisol concentrations, and unconjugated urinary cortisol excretion, these findings suggest that the relationship between these other aspects of cortisol metabolism and insulin sensitivity is relatively weak.
Previously it has been shown that obese individuals [12] and overweight people with type 2 diabetes [31] with fatty liver have higher 24-h urinary free cortisol excretion with impaired dexamethasone suppression of cortisol compared with control subjects. However, the effect of fatty liver per se on cortisol metabolism is not differentiated from the effect mediated by obesity in these studies. Moreover, the value of the dexamethasone suppression test is limited in these circumstances [32]. Subjects with decreased dexamethasone suppression have more rapid dexamethasone clearance [33], the rate of dexamethasone metabolism is increased in people in whom the suppression of cortisol during the dexamethasone suppression test is decreased [32, 33], and the rate of metabolic clearance of dexamethasone is higher in obese people than in people of normal weight [34]. Furthermore, the capacity of the liver to metabolise dexamethasone is related to liver function parameters that are known to be abnormal in NAFLD [35]. Also, because synthetic steroid hormones such as dexamethasone are metabolised in the liver before being eliminated [36], it is possible that a disorder that adversely affects liver function, such as NAFLD, may also affect dexamethasone metabolism and influence the suppression of cortisol during this test. For these reasons we chose not to undertake dexamethasone suppression tests and instead chose to assess whole-body cortisol clearance by assessing clearance of an isotope of cortisol from the plasma.
CBG concentration was inversely related to cortisol clearance and positively associated with insulin sensitivity. It is plausible that the decreased concentrations of CBG in subjects with fatty liver contributed to increased cortisol clearance in these subjects. The relationship between CBG concentrations and insulin sensitivity remains unclear. Some investigators have failed to show an association between CBG concentrations and insulin sensitivity [37]. In contrast, other authors have demonstrated an association. Recently, a negative correlation between CBG concentration and BMI, waist to hip ratio and insulin resistance in normal healthy subjects has been demonstrated [38], and it was also shown that plasma adiponectin was associated with serum CBG in men (r = 0.26, p = 0.03) [39]. Furthermore, CBG concentration has now been shown to be inversely related to measures of inflammation [38]. Therefore, the CBG concentration may be a marker of an acute-phase response rather than directly reflecting insulin sensitivity. Since it is now widely accepted that insulin resistance is associated with low-grade inflammation in the metabolic syndrome, a decrease in CBG concentrations may reflect or be causally related to the inflammatory state. In our study there were correlation coefficients (albeit not statistically significant) similar to those in the above study [39] between CBG and: (1) adiponectin; (2) TNF-α; (3) IL-6; and (4) IL-1β (r = 0.32, p = 0.1 for adiponectin; r = −0.3, p = 0.17 for TNF-α; r = −0.31, p = 0.15 for IL-6; and r = −0.23, p = 0.29 for IL-1β). Consequently, taking our results and the published literature into consideration, we suggest that further studies are needed to explain whether inflammation plays any role in helping explain the strong association between cortisol clearance and insulin sensitivity that we have observed.
What is the mechanism underlying increased cortisol clearance with insulin resistance? In obesity it is known that the tissue-specific metabolic activation of the glucocorticoid precursor (cortisone in humans) to the active hormone cortisol is carried out by type 1 11β-hydroxysteroid dehydrogenase (11β-HSD1) in both liver and fat. There is evidence of differential activity of 11β-HSD1 in key insulin-sensitive tissues such as liver and fat in obesity. In such individuals there is decreased activity of 11β-HSD1 in liver and increased activity in fat [40]. Also, we and others have shown that skeletal muscle has 11β-HSD1 activity [41–43] and can generate cortisol from cortisone, suggesting that all three key insulin-sensitive tissues have the capacity to generate cortisol. It is also possible that individuals differ in their ability to generate cortisol from cortisone in each of these three tissues. Furthermore, it is plausible that increased tissue-mediated cortisol clearance with decreased insulin sensitivity reflects a resetting of the HPA axis in insulin-resistant states, with no compensatory increase in cortisol production to overcome the increased cortisol clearance. The net consequence of such an effect might be a decrease in the 09.00 h cortisol concentration. In support of this suggestion, we have recently observed a decrease in 09.00 h cortisol concentration in South Asians compared with white northern Europeans [44]. In interpreting these data and the presented results showing increased cortisol clearance, it is plausible that the HPA axis might be reset in insulin-resistant individuals, and fail to respond to increased cortisol clearance, by increasing cortisol production via increased HPA axis activity. The net effect of such resetting of the HPA axis might be a flatter, lower 24-h cortisol profile. Such a profile is in keeping with the original findings of Bjorntorp et al. [45], who measured 24-h salivary cortisol concentrations, and described an association between features of the metabolic syndrome and a flat rigid cortisol day curve, with poor feedback control. Thus, we suggest that our study now emphasises the need to undertake cortisol turnover studies to understand fully the relationship between cortisol metabolism and insulin resistance.
In summary, we have found a strong association between cortisol clearance and insulin sensitivity that is independent of the amounts of total or truncal body fat. We have also shown that the relationship between fatty liver and insulin sensitivity is mediated in part by fatness and in part by increased cortisol clearance.
Abbreviations
- CBG:
-
corticosteroid-binding globulin
- DEXA:
-
dual-energy X-ray absorptiometry
- HPA:
-
hypothalamic–pituitary–adrenal
- MRI:
-
magnetic resonance imaging
- NAFLD:
-
non-alcoholic fatty liver disease
References
Strain GW, Zumoff B, Kream J, Strain JJ, Levin J, Fukushima D (1982) Sex difference in the influence of obesity on the 24 hr mean plasma concentration of cortisol. Metabolism 31:209–212
Jessop DS, Dallman MF, Fleming D, Lightman SL (2001) Resistance to glucocorticoid feedback in obesity. J Clin Endocrinol Metab 86:4109–4114
Purnell JQ, Brandon DD, Isabelle LM, Loriaux DL, Samuels MH (2004) Association of 24-hour cortisol production rates, cortisol-binding globulin, and plasma-free cortisol levels with body composition, leptin levels, and aging in adult men and women. J Clin Endocrinol Metab 89:281–287
Andrew R, Phillips DI, Walker BR (1998) Obesity and gender influence cortisol secretion and metabolism in man. J Clin Endocrinol Metab 83:1806–1809
Lottenberg SA, Giannella-Neto D, Derendorf H et al (1998) Effect of fat distribution on the pharmacokinetics of cortisol in obesity. Int J Clin Pharmacol Ther 36:501–505
Reynolds RM, Walker BR, Syddall HE et al (2001) Altered control of cortisol secretion in adult men with low birth weight and cardiovascular risk factors. J Clin Endocrinol Metab 86:245–250
Walker BR, Soderberg S, Lindahl B, Olsson T (2000) Independent effects of obesity and cortisol in predicting cardiovascular risk factors in men and women. J Intern Med 247:198–204
Ahmed MH, Byrne CD (2005) Non alcoholic steatohepatitis. In: Byrne CD, Wild SH (eds) The metabolic syndrome, 1st edn. Wiley, Chichester, pp 279–303
Angulo P (2002) Nonalcoholic fatty liver disease. N Engl J Med 346:1221–1231
Holt HB, Wild SH, Wood PJ et al (2006) Non-esterified fatty acid concentrations are independently associated with hepatic steatosis in obese subjects. Diabetologia 49:141–148
Utzschneider KM, Kahn SE (2006) The role of insulin resistance in non-alcoholic fatty liver disease. J Clin Endocrinol Metab 91:4753–4761
Zoppini G, Targher G, Venturi C, Zamboni C, Muggeo M (2004) Relationship of nonalcoholic hepatic steatosis to overnight low-dose dexamethasone suppression test in obese individuals. Clin Endocrinol (Oxf) 61:711–715
Saruc M, Yuceyar H, Ayhan S, Turkel N, Tuzcuoglu I, Can M (2003) The association of dehydroepiandrosterone, obesity, waist-hip ratio and insulin resistance with fatty liver in postmenopausal women—a hyperinsulinemic euglycemic insulin clamp study. Hepatogastroenterology 50:771–774
Westerbacka J, Yki-Jarvinen H, Vehkavaara S et al (2003) Body fat distribution and cortisol metabolism in healthy men: enhanced 5beta-reductase and lower cortisol/cortisone metabolite ratios in men with fatty liver. J Clin Endocrinol Metab 88:4924–4931
Targher G, Bertolini L, Rodella S, Zoppini G, Zenari L, Falezza G (2006) Associations between liver histology and cortisol secretion in subjects with nonalcoholic fatty liver disease. Clin Endocrinol (Oxf) 64:337–341
DeFronzo RA, Tobin JD, Andres R (1979) Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 237:E214–E223
Powrie JK, Smith GD, Hennessy TR et al (1992) Incomplete suppression of hepatic glucose production in non-insulin dependent diabetes mellitus measured with [6,6-2H2]glucose enriched glucose infusion during hyperinsulinaemic-euglycaemic clamps. Eur J Clin Invest 22:244–253
Finegood DT, Bergman RN (1983) Optimal segments: a method for smoothing tracer data to calculate metabolic fluxes. Am J Physiol 244:E472–E479
Steele R (1959) Influences of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci 82:420–430
Finegood DT, Bergman RN, Vranic M (1988) Modeling error and apparent isotope discrimination confound estimation of endogenous glucose production during euglycemic glucose clamps. Diabetes 37:1025–1034
Belfiore F, Iannello S, Volpicelli G (1998) Insulin sensitivity indices calculated from basal and OGTT-induced insulin, glucose, and FFA levels. Mol Genet Metab 63:134–141
Moore A, Aitken R, Burke C et al (1985) Cortisol assays: guidelines for the provision of a clinical biochemistry service. Ann Clin Biochem 22:435–454
Jerjes WK, Cleare AJ, Peters TJ, Taylor NF (2006) Circadian rhythm of urinary steroid metabolites. Ann Clin Biochem 43:287–294
Levey AS, Bosch JP, Lewis JB, Greene T, Rogers N, Roth D (1999) A more accurate method to estimate glomerular filtration rate from serum creatinine: a new prediction equation. Modification of Diet in Renal Disease Study Group. Ann Intern Med 130:461–470
Bright GM (1995) Corticosteroid-binding globulin influences kinetic parameters of plasma cortisol transport and clearance. J Clin Endocrinol Metab 80:770–775
Tait JF (1963) Review: the use of isotopic steroids for the measurement of production rates in vivo. J Clin Endocrinol Metab 23:1285–1297
Kleinbaum D, Kupper L, Muller K, Nizam A (1998) Multiple regression analysis: general considerations. In: Applied regression analysis and other multivariate methods. Duxberry, Pacific Grove, CA, pp 237–245
Clasey JL, Bouchard C, Teates CD et al (1999) The use of anthropometric and dual-energy X-ray absorptiometry (DXA) measures to estimate total abdominal and abdominal visceral fat in men and women. Obes Res 7:256–264
Aldhahi W, Mun E, Goldfine AB (2004) Portal and peripheral cortisol levels in obese humans. Diabetologia 47:833–836
Saverymuttu SH, Joseph AE, Maxwell JD (1986) Ultrasound scanning in the detection of hepatic fibrosis and steatosis. Br Med J (Clin Res Ed) 292:13–15
Targher G, Bertolini L, Zoppini G, Zenari L, Falezza G (2005) Relationship of non-alcoholic hepatic steatosis to cortisol secretion in diet-controlled type 2 diabetic patients. Diabet Med 22:1146–1150
Pasquali R, Ambrosi B, Armanini D et al (2002) Cortisol and ACTH response to oral dexamethasone in obesity and effects of sex, body fat distribution, and dexamethasone concentrations: a dose–response study. J Clin Endocrinol Metab 87:166–175
Cassidy F, Ritchie JC, Verghese K, Carroll BJ (2000) Dexamethasone metabolism in dexamethasone suppression test suppressors and nonsuppressors. Biol Psychiatry 47:677–680
Tsuei SE, Moore RG, Ashley JJ, McBride WG (1979) Disposition of synthetic glucocorticoids. I. Pharmacokinetics of dexamethasone in healthy adults. J Pharmacokinet Biopharm 7:249–264
Huizenga NA, Koper JW, de Lange P et al (1998) Interperson variability but intraperson stability of baseline plasma cortisol concentrations, and its relation to feedback sensitivity of the hypothalamo-pituitary-adrenal axis to a low dose of dexamethasone in elderly individuals. J Clin Endocrinol Metab 83:47–54
Kutemeyer S, Schurmeyer TH, von zur Muhlen A (1994) Effect of liver damage on the pharmacokinetics of dexamethasone. Eur J Endocrinol 131:594–597
Lewis JG, Shand BI, Elder PA, Scott RS (2004) Plasma sex hormone-binding globulin rather than corticosteroid-binding globulin is a marker of insulin resistance in obese adult males. Diabetes Obes Metab 6:259–263
Fernandez-Real JM, Pugeat M, Grasa M et al (2002) Serum corticosteroid-binding globulin concentration and insulin resistance syndrome: a population study. J Clin Endocrinol Metab 87:4686–4690
Fernandez-Real JM, Pugeat M, Lopez-Bermejo A, Bornet H, Ricart W (2005) Corticosteroid-binding globulin affects the relationship between circulating adiponectin and cortisol in men and women. Metabolism 54:584–589
Wake DJ, Walker BR (2006) Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 in obesity. Endocrine 29:101–108
Whorwood CB, Donovan SJ, Wood PJ, Phillips DI (2001) Regulation of glucocorticoid receptor alpha and beta isoforms and type I 11beta-hydroxysteroid dehydrogenase expression in human skeletal muscle cells: a key role in the pathogenesis of insulin resistance? J Clin Endocrinol Metab 86:2296–2308
Reynolds RM, Chapman KE, Seckl JR, Walker BR, McKeigue PM, Lithell HO (2002) Skeletal muscle glucocorticoid receptor density and insulin resistance. JAMA 287:2505–2506
Whorwood CB, Donovan SJ, Flanagan D, Phillips DI, Byrne CD (2002) Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes 51:1066–1075
Reynolds RM, Fischbacher C, Bhopal R et al (2006) Differences in cortisol concentrations in South Asian and European men living in the United Kingdom. Clin Endocrinol (Oxf) 64:530–534
Bjorntorp P, Holm G, Rosmond R (1999) Hypothalamic arousal, insulin resistance and type 2 diabetes mellitus. Diabet Med 16:373–383
Acknowledgements
We would like to thank the staff of the Wellcome Trust Clinical Research Facility for their help with this study. We would like to thank C. Wang for her help with the assays, L. England for her help with this manuscript and N. Englyst and A. Ward for their advice. We are grateful to M. Hanna for performing the ultrasonography and scoring the liver scans. The work was funded by grant support from the Wellcome Trust to C.B., from Wessex Deanery, and from NHS Research and Development funding.
Duality of interest
There are no conflicts of interest for all authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Holt, H.B., Wild, S.H., Postle, A.D. et al. Cortisol clearance and associations with insulin sensitivity, body fat and fatty liver in middle-aged men. Diabetologia 50, 1024–1032 (2007). https://doi.org/10.1007/s00125-007-0629-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-007-0629-9