
Abstract
Mapping chromosome regions responsible for quantitative phenotypic variation in recombinant populations provides an effective means to characterize the genetic basis of complex traits. We conducted a quantitative trait loci (QTL) analysis of 150 rice recombinant inbred lines (RILs) derived from a cross between two cultivars, Oryza sativa ssp. indica cv. 93-11 and Oryza sativa ssp. japonica cv. Nipponbare. The RILs were genotyped through next-generation sequencing, which accurately determined the recombination breakpoints and provided a new type of genetic markers, recombination bins, for QTL analysis. We detected 49 QTL with phenotypic effect ranging from 3.2 to 46.0% for 14 agronomics traits. Five QTL of relatively large effect (14.6–46.0%) were located on small genomic regions, where strong candidate genes were found. The analysis using sequencing-based genotyping thus offers a powerful solution to map QTL with high resolution. Moreover, the RILs developed in this study serve as an excellent system for mapping and studying genetic basis of agricultural and biological traits of rice.
Similar content being viewed by others

Introduction
Genetic variation of a complex trait is usually controlled by multiple loci. When studied in a recombinant population, the trait typically varies in a continuous manner. The use of molecular genetic markers decades ago enabled the detection of chromosomal regions harboring quantitative trait loci (QTL). Since then, the number of studies to map QTL has increased rapidly, fueled primarily by interests to identify the genetic control of agriculturally, medically, and ecologically important traits (Tanksley 1993; Lander and Schork 1994; Mackay 2001; Mauricio 2001). Advances in molecular biology and genomic techniques have then made it possible to narrow down a QTL to a few or even a single candidate gene (Doebley et al. 1997; Frary et al. 2000; Yano et al. 2000; Grisart et al. 2002). The cloning of QTL and identification of causative mutations have opened an avenue to unlock the genetic basis of complex phenotypic variation.
However, cloning QTL remains technically challenging. It either requires the development of near-isogenic lines (NILs) through repeatedly backcrossing with one of the mapping parents (Ashikari et al. 2005; Konishi et al. 2006; Song et al. 2007; Jin et al. 2008; Shomura et al. 2008; Xue et al. 2008; Li et al. 2006) or additional samples of natural variants for association of phenotype and candidate genes (Grisart et al. 2002; Van Laere et al. 2003; Sutter et al. 2007; Harjes et al. 2008). Positional cloning using NILs is time-consuming and labor-intensive because it takes a few generations of backcrossing to make NILs and thousands of recombinants to fine map the candidate genes. It could be prohibitively tedious and prolonged for organisms with relatively long life cycles or relatively few offspring from crosses. With regard to the other cloning strategy involving association analysis, the difficulty arises as it often relies on the presence of candidate genes with known function near the QTL.
The whole-genome sequencing approach takes advantage of a recently developed genotyping method that uses single nucleotide polymorphisms (SNPs) detected from whole-genome sequencing of a mapping population (Huang et al. 2009). This approach could substantially reduce the amount of time and effort required for QTL mapping. The SNPs were evaluated in sliding windows to generate recombination maps for the individuals. The maps were then aligned and used to define recombination bins for the entire population. Recombination bins can serve as a new and effective type of genetic markers for QTL analysis. It is different from conventional molecular markers, such as random amplified polymorphic DNA (RAPD), restriction fragment length polymorphisms (RFLPs), insertion–deletion markers (In/Del), and simple sequence repeat (SSR), which often have uneven distribution and low density on whole genome and need more time on genotyping. The bins, which presumably capture all recombination events in the population, provide availably abundant markers based on dense SNPs for detailed genome-wide trait analysis.
In this study, we reported high-resolution QTL mapping through sequencing-based genotyping of 150 rice recombinant inbred lines (RILs). The population was developed from a cross between two rice cultivars with genome sequences, Oryza sativa ssp. japonica cv. Nipponbare and Oryza sativa spp. indica cv. 93-11 (Goff et al. 2002; Yu et al. 2002; International Rice Genome Sequencing Project 2005). We identified 49 QTL within relatively small genomic regions for 14 agronomic traits. With a relatively high mapping resolution, we were able to identify the candidate genes for some QTL of large or moderate effect. The new genotyping method thus greatly improved the resolution and precision of QTL mapping for complex traits.
Materials and methods
Mapping population
The rice mapping population of 150 RILs was derived by single-seed descents from a cross between Oryza sativa ssp. indica cv. 93-11 and Oryza sativa ssp. japonica cv. Nipponbare. The population was developed in the experimental fields at China National Rice Research Institute in Hangzhou, Zhejiang Province, and Sanya, Hainan Province. After ten generations of self-fertilization following the initial cross, DNAs of the F11 RILs were isolated for genotyping. Phenotyping was conducted in the Hangzhou field (N 30.32°, E 120.12°) from May to October, 2008, and in the laboratory following harvest.
Phenotyping
Of 18 individuals of each RIL and parent grown in the field, 5 plants were randomly chosen for phenotyping. A total of 14 traits were evaluated. Traits measured directly in the field include heading date, culm diameter, plant height, flag leaf length and flag leaf width, tiller angle, tiller number, panicle length, and awn length. Traits measured in the laboratory following harvest include grain length, grain width, grain thickness, grain weight, and spikelet number per panicle (Table S1).
Heading date was recorded as days from sowing to time when inflorescences had emerged above the flag leaf sheath for more than half of the individuals of line. Culm diameter, plant height, flag leaf length and width, and tiller angle were evaluated when panicles fully emerged. Culm diameter was measured at the thickest location of the third tiller node from the root; tiller angle was scored on a 1–6 scale (1, <10° between tiller and vertical; 6, >45°). Plant height was measured from the soil surface to the apex of the tallest panicle. On the main tiller, flag leaf length was measured from leaf blade and sheath boundary to the leaf apex; flag leaf width was measured at the widest location of the leaf. Tiller number, panicle length, and awn length were evaluated when grains fully matured. All flowered tillers of an individual were counted, the longest panicle was measured in length, and five grains located on the top of this panicle were chosen for measuring awn length. The total number of spikelets produced on the main tiller was counted.
The grain related traits were measured in the laboratory after grains were detached from panicles and awns were removed from the grains. For the sampled panicles of an individual, grains were mixed and 10 grains were randomly sampled for phenotyping. Grain length, width, and thickness were recorded at the maximal values for each grain using an electronic digital caliper. Grain weight was initially obtained by weighing a total of 200 grains, which was then converted to 1,000-grain weight, a scale commonly used for yield evaluation.
Genotyping, linkage map, and QTL analysis
A high-throughput genotyping method was previously developed and tested using these 150 rice RILs (Huang et al. 2009). The RILs were genotyped based on SNPs generated from the whole-genome resequencing. A recombination map was constructed for each RIL. The recombination maps were aligned to determine recombination bins across the entire population with the minimal bin length of 100 kb adopted. Resulting bins were then treated as a genetic marker for linkage map construction using MAPMAKER/EXP version 3.0b (Lander et al. 1987).
Using this linkage map and phenotypic values, QTL analysis was conducted with the composite interval mapping (CIM) implemented in software Windows QTL Cartographer V2.5 (Wang et al. 2007) (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm). The CIM analysis was run using Model 6 with forward and backward stepwise regression, a window size of 10 cM, and a step size of 2 cM. Experiment-wide significance (P < 0.05) thresholds for QTL detection were determined with 1,000 permutations. The location of a QTL was described according to its LOD peak location and the surrounding region with 95% confidence interval calculated using WinQTLCart. The epistasis between QTL was estimated using R/qtl in the R package (http://www.rqtl.org) (Broman et al. 2003).
Simulation schemes
To evaluate the effect of marker density, two sets of markers with different density were simulated for QTL analysis. For the set with low marker density, 238 locations evenly distributed in the rice genome were designated with the density of 1 marker per 1.6 Mb based on physical position. Then each location was treated as one simulated marker, and the genotype of the marker was deduced from genotype of the recombination bin where the marker was located. In this way, genotypes of 150 individuals with a total of 238 simulated markers were obtained (Table S2). The set with high-marker density was simulated in the same way. The density was 1 marker per 164 kb, which generated a total of 2,330 markers.
To evaluate the effect of population size, 50 and 100 lines were randomly sampled from 150 RILs five times for QTL analysis, respectively. Moreover, genotypes and phenotypes were simulated five times for each population size (from 50 to 500 individuals) for QTL analysis using the simulation module in the software WinQTLCart. In the simulation, chromosome number and marker position were imported according to 2,334 bins, and the QTL information was imported based on the 49 QTL mapped using the 150 RILs.
The way to construct genetic map and QTL analysis using the simulated markers and populations was the same as that for the 150 RILs.
Results
Phenotypic variation
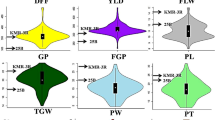
Phenotypic variation of the rice RILs and parents is illustrated in Fig. 1 and supplemental Fig. S1. Of the 14 traits evaluated, 10 showed significant differences between the indica and japonica mapping parental lines and 2 (culm diameter and tiller number) were not significantly different between the parental lines, while the significance level was not determined for heading date or tiller angle (Table S1). All traits showed transgressive segregation in the RIL population (Fig. 1).

Variation of phenotypic traits in RILs. Mean and standard deviation of the parents are indicated at the top of each histogram, with i and j representing O. sativa ssp. indica cv. 93-11 and O. sativa ssp. japonica cv. Nipponbare, respectively
The correlation of trait variation is illustrated in Fig. 2. Significantly positive correlation is found among nine traits (green shading), awn length, grain length, culm diameter, spikelet number, plant height, panicle length, flag leaf length, flag leaf width, and heading date. This group of nine traits shows significantly negative correlation with other four traits (yellow shading), tiller angle, tiller number, grain width, and grain thickness. Specifically, grain thickness is negatively correlated with spikelet number, panicle length, flag leaf length, and heading date; grain width is negatively correlated with awn length, grain length, culm diameter, spikelet number, and panicle length; tiller number is negatively correlated with culm diameter, spikelet number, panicle length, plant height, flag leaf length, and flag leaf width; tiller angle is negatively correlated with flag leaf length and width. Between these four traits, grain thickness and grain width are significantly positive correlation. The remaining trait (purple shading), 1,000-grain weight, was positively correlated with grain thickness and grain width, and shows both positive and negative correlations with the first group of nine traits. It is positively correlated with awn length, grain length, culm diameter, panicle length, and plant height, and negatively correlated with spikelet number and heading date.

The correlation of trait variation. Blue and red lines indicated positive and negative correlations, respectively. Solid lines P < 0.01; dotted lines 0.01 < P < 0.05. AL awn length, GL grain length, CD culm diameter, SN spikelet number, PL panicle length, PH plant height, FLL flag leaf length, FLW flag leaf width, HD heading date, TA tiller angle, TN tiller number, GW grain width, GT grain thickness, TGW 1,000-grain weight
Linkage map of recombination bins
A linkage map was constructed using 2,334 recombination bins which was obtained from the whole-genome resequencing of the 150 RILs (Huang et al. 2009), which resulted in a total genetic distance of 1,539.5 cM with an average interval of 0.66 cM between adjacent bins. For each chromosome, the average genetic distance between adjacent bins ranging 0.66–0.82 cM, with the maximal distance between 2.1 and 8.3 cM (Table S3).
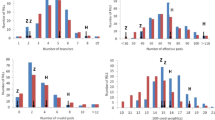
The linkage map constructed from the bins is compared with a map generated from an F2 population of 186 individuals derived from a cross between japonica cv. Nipponbare and indica cv. Kasalath (Harushima et al. 1998) (Table S3). This represents the rice linkage map covered with the largest number of conventional molecular markers reported to date, where we found a total of 3,235 genetic markers including RFLP, RAPD, and STS from the most updated version (http://www.gramene.org/db/cmap/map_set_info?map_set_acc=jrgp-rflp-2000). The total genetic distance of the 12 chromosomes of these two maps is very close. The average genetic distance between adjacent bins with greater than zero distance is 0.72 cM on our map, smaller than the average of 1.03 cM for the conventional markers. The maximal genetic distance between adjacent markers is 8.3 and 15.6 cM on the bin and conventional maps, respectively. Furthermore, on the map with conventional markers, more than half (53.7%) of the adjacent markers have genetic distance of 0, whereas only about 7.9% of adjacent bins had zero genetic distance as calculated by MAPMAKER. In addition, 76.6% of adjacent bins had genetic distance between 0.1–1 cM, whereas 31.6% of adjacent markers have this level of resolution on the conventional map (Fig. 3; Table S4). Therefore, the map with recombination bins has well-distributed linkage distance and higher resolution than the conventional map.

Comparison of chromosomal coverage between bins, high-density simulated markers, and conventional molecular markers. Bars indicate the frequency of genetic distance between adjacent markers on the linkage maps. White bars bin markers from this study; gray bars, simulated markers from this study; black bars conventional molecular markers from a previously studied rice F2 population
QTL analysis
The LOD thresholds for QTL calling were estimated from the permutation test and ranged from 2.85 for tiller number to 3.48 for flag leaf width. Based on these thresholds, a total of 40 QTL were called for the 14 traits, with phenotypic effect (R 2) of the QTL ranging 4.3–46.0% (Table 1). Considering the power of QTL detection with 150 RILs, we also reported QTL with LOD value higher than 3.0. This gives nine additional QTL, with phenotypic effect ranging 3.2–7.0% (Table 1). Thus a total of 49 QTL are detected on 12 rice chromosomes, with 1–5 QTL detected for each trait (Fig. 4). The region of each QTL identified in this study was based on the 95% confidence interval (CI) calculated using WinQTLCart (Wang et al. 2007). Of them, QTL that explained more than 10% of phenotypic effects were defined as major-effect QTL here. We totally identified 10 major-effect QTL, including qTA-9, qPH-1, qFLW-4, qGL-3, qGW-5, qAL-1, qAL-3, qPH-2, qHD-3, and qCD-2.

Chromosomal locations of detected QTL. Twelve rice chromosomes are numbered, with centromeres indicated by open circles. Positions of 2,334 bins used as genetic markers are indicated by horizontal lines spaced proportionally according to their genetic distance on the linkage map. Arrow heads indicate LOD peak locations of QTL. Black arrow heads QTL with LOD values above the threshold determined by permutation tests; gray arrow heads QTL with LOD values above 3.0 but below the permutation threshold. For QTL abbreviations, see Table 1
We searched the literatures and database for previously identified QTL from mapping populations also derived from crosses between indica and japonica cultivars. QTL detected in this study were compared with those previously identified by physically locations that could be clearly determined. In this condition, 18 of our QTL for 11 traits fell into the chromosomal regions containing the QTL identified in the previous studies (Table 2), including the top 5 large-effects QTL and another major-effect QTL (qHD-3). The remaining 31 were not found. These new QTL include two of the three QTL for culm diameter, two of the four QTL for plant height, two of the three QTL for flag leaf length, three of the four QTL for flag leaf width, two of the three QTL for tiller angle, one QTL for tiller number, four of the five QTL for panicle length, three of the four QTL for grain length, two of the five QTL for grain width, one of the four QTL for grain weight, and one of the four QTL for spikelet number. Almost all traits in this study have novel QTL except heading date. In addition, among 31 newly detected QTL, 4 QTL above mentioned with large effect have not been reported to date which is involved with awn length, plant height, and culm diameter.
We found that the LOD peak of a QTL always overlapped with the genetic position of a bin marker. This location of the QTL was then pinpointed on the chromosome (Fig. 4). The 95% confidence interval of the QTL location was determined (Table 1). If 95% confidence intervals of the two QTL were overlapped, these QTL were defined to be co-localized. A total of seven groups of co-localized QTL are identified, including (1) qFLL-2 and qGW-2b on chromosome 2; (2) qCD-2, qPH-2, and qPL-2 on chromosome 2; (3) qAL-3, qGL-3 and qGW-3 on chromosome 3; (4) qGT-5 and qGW-5 on chromosome 5; (5) qTN-5 and qFLL-5 on chromosome 5; (6) qTA-6 and qGL-6 on chromosome 6; and (7) qGW-6, qGT-6, and qSN-6 on chromosome 6.
Epistasis analysis detected significant interaction between nine pairs of QTL for six traits (Table 3). These include one pair each for flag leaf length, flag leaf width, and awn length, and two pairs each for grain length, 1,000-grain weight, and spikelet number. Because only additive effect of QTL can be estimated from the RIL population, all interactions are additive–additive epistasis with effect sizes ranging 1.09–4.87%.
Candidate genes
With a relatively high mapping resolution, the small genomic regions of top five large-effects QTL are overlapped with the locations of five genes reported in previous studies (Fig. 5). For the QTL of the largest effect on tiller angle, qTA-9 (R 2 = 46.0%) was located in the region which ranges from 21.3 to 21.7 Mb on chromosome 9. A strong candidate gene, TAC1, that controls tiller angle was previously cloned (Yu et al. 2007) and is located in this region. For plant height, the major QTL, qPH-1 (R 2 = 30.0%) has its LOD peak in the region of 600 kb on chromosome 1. On the same location, SD1, a gene responsible for rice semi-dwarfness and known as the rice green revolution gene (Sasaki et al. 2002), is located. The major QTL for flag leaf width, qFLW-4 (R 2 = 16.4%), is mapped to the region which ranges from 31.3 to 31.8 Mb on chromosome 4, while a gene controlling leaf width, Nal1, was found in it (Qi et al. 2008). The QTL of largest effect on grain length, qGL-3 (R 2 = 16.1%), has its LOD peak in the region of 700 kb on chromosome 3, which contains GS3, a gene controlling grain shape, was cloned (Fan et al. 2006). Finally, the major QTL for grain width, qGW-5 (R 2 = 14.6%), is located in the region of 400 kb on chromosome 5, which is overlapped with the location where qSW5 that controls grain width was previously cloned (Shomura et al. 2008).

Precise locations of five QTL and candidate genes. Curves indicate chromosomal locations and LOD values of detected QTL. Names and phenotypic effect (R 2) of the QTL are indicated. Recombination bins near the LOD peaks of the QTL are illustrated as horizontal bars with their numbers labeled inside. Shaded bin overlaps the LOD peak of a QTL. The portion of a curve and corresponding bins defining the 95% confidence interval of a QTL are bracketed by two lines. The relative physical position of a candidate gene in the bin is indicated by an arrow head
Effects of genotype resolution and population size on QTL mapping
In order to evaluate the effect of high-throughput genotyping method on QTL mapping resolution, we designed a set of low density (238) and a set of high density (2,330) markers evenly distributed in the rice genome (Table S2) for QTL analysis. On the linkage map constructed by high-density simulated markers, which is comparably dense to bins, 755 pairs of adjacent markers (32.6%) have genetic distance of zero, which was more than the linkage map constructed by bins (Fig. 3; Table S4). It was likely because that these simulated markers were set to specific locations, such as regions around the centromeres and regions with low-recombination rates. Then 29 and 43 QTL were mapped using these low- and high-density simulated markers, respectively, which overlapped with QTL identified using sequencing-based genotyping dataset (Table S5). The 95% confidence intervals (CI) of the latter set of QTL were significantly smaller than those of two former sets of QTL (paired-sampled t test, both P < 0.001; and independent t test, P < 0.001 and P = 0.037, respectively; Table S5). It suggested that QTL mapping resolution using the high-throughput genotyping method was increased significantly.
Moreover, in order to evaluate the effect of population size on QTL mapping resolution using the high-throughput genotyping method, two schemes were employed. In the scheme 1, 50 and 100 RILs were randomly sampled from original 150 RILs five times, respectively. We observed that the possibility with which five major QTL can be detected dropped to 76% (19 of 25) when population size decreased to be 100, and the possibility dropped to 44% (11 of 25) when the size got to be 50 (Table S6). It suggested that the smaller population sizes affected the sensitivity of QTL mapping dramatically. In the scheme 2, various sizes of population size (from 50 to 500 individuals) were simulated by WinQTLCart. On average, the 95% confidence interval of five major QTL was changed from 6.7 to 1.4 cM (the mean value of five parallel simulation) when population size was transformed from 50 to 500, and their standard deviation was changed from 3.2 to 0.7 (Table S7, Fig. S2). It suggested that lager population size coupled with high-resolution genotype information could further increase QTL mapping resolution.
Discussion
Relationship of mapped QTL and previously identified genes
We found that there were strong candidate genes for the top five largest-effect QTL. For example, the known TAC1 gene (Yu et al. 2007) was found to be located in the region of qTA-9 on chromosome 9. Comparative genome sequencing analysis revealed that Nipponbare shared the same mutation with all japonica cultivars which cause small tiller angles than indica rice. This strongly supports the candidacy of TAC1 for qTA-9. The same situation happened to other four QTL, qPH-1, qFLW-4, qGL-3, and qGW-5, which contains SD1, Nal1, GS3, and qSW5, respectively (Sasaki et al. 2002; Qi et al. 2008; Fan et al. 2006; Shomura et al. 2008). The detailed information for the strong candidate genes involved in five major QTL is shown in Fig. S3. Of the candidate genes for these QTL, four were cloned from QTL identified from other rice cultivars. They are likely the genes with widespread large effect on natural variation among rice cultivars. Besides these genes, few others have been cloned for the natural variation of complex traits in rice.
There was no such strong evidence for the remaining QTL. However, we found that some candidate genes might support some of them, such as qHD-3. OsSOC1 that activate flowering in rice was found to be located in the region of qHD-3 for heading date in this study (Tadege et al. 2003). OsSOC1, which encodes a MADS domain protein, is the ortholog of the Arabidopsis gene AtSOC1. AtSOC1 and OsSOC1 show a 97% amino acid similarity in their MADS domain (Tadege et al. 2003).
Co-localization of mapped QTL
A total of seven co-localized QTL groups were identified among 49 QTL. We found that five groups of them underlie significant correlation between ten pairs of traits. Among them, five pairs of traits have significantly positive correlation, including CD and PH, CD and PL, PH and PL, AL and GL, and GW and GT; the other five pairs have significantly negative correlation, including AL and GW, GL and GW, TN and FLL, GW and SN, and GT and SN. These co-localizations suggested that pleiotropic effect or linkage of the genes is likely to be responsible for the trait correlation. With the data in the present form, however, we could not distinguish these two alternatives. Nevertheless, not all correlations can be explained by QTL co-localization. This could be due to the effect of undetected QTL or reasons other than pleiotropy or linkage.
Genotype resolution and population size affect QTL mapping resolution
QTL mapping resolution, determined by the size of confidence interval of QTL, depends on the population size and marker density (Visscher et al. 1996; Da et al. 2000). In general, a gain of information, provided by a greater population size and a more markers density, resulted in smaller intervals (Bennewitz et al. 2002). The former determines the number of recombination events, while the latter determines the resolution of recombination breakpoints.
Development and exploitation of additional markers is one of the strategies to increase QTL mapping resolution (Gautier et al. 2006; Liu et al. 2008). In this study, QTL mapping resolution using the sequencing-based genotyping method (Huang et al. 2009) is significantly increased by comparing with those using simulated markers. Based on the comparison among linkage maps generated from bins, simulated markers, and conventional molecular markers, bins detect a larger number of recombination events with high accuracy. Evidently, recombination bins are superior to conventional molecular markers in genome coverage. Unlike conventional molecular markers, recombination bins defined by SNPs detected from whole-genome sequencing allow the entire genome to be evenly evaluated and accurately represent recombination events across the RILs, almost each one of which is available in QTL analysis. Although conventional markers are based on distribution of nucleotide polymorphism on the whole genome, a significant proportion of them is unavailable when their linkage distance are zero. This is also supported by the fact that physical sizes and regions surrounding centromeres have the largest bins and lowest recombination rates (Fig. S4). It suggested that the conceptual and technical advances in genetic markers have improved the accuracy of QTL mapping.
There was some difference in detected QTLs between genotyped 150 RILs and simulations. The possible basis is the specificity of composite interval mapping, which uses other markers to control genetic background when testing for the putative QTL (Kao et al. 1999).
Furthermore, population size plays a key role in QTL mapping resolution. In our study, QTL analysis using simulated populations showed that QTL mapping resolution increased with increase in population size, which is consistent with the simulation study by Visscher et al. (1996). Especially, increasing population size was more efficient than increasing marker density in QTL analysis when the individual number of population was less than 150 in this study (Table S6).
For this study, it is the advantage of the sequencing-based genotyping method that allows mapping population to be expanded and more deeply genotyped easily. Additional RILs derived from the same cross can be genotyped to increase the mapping population size. The RILs can be sequenced again at higher genome coverage to add more SNPs and detect additional recombination events. These two efforts together can cut the recombination bins into smaller sizes, which should help resolve QTL in a finer scale. The RIL population between rice cultivars, Nipponbare and 93-11, provides an excellent system for studying genetic basis of complex traits. Especially, individuals in RILs as permanent segregating populations are homozygous at all loci throughout the genome, and individuals within lines are highly homogeneous and thus can be used for repeated trials both over times and locations (Mei et al. 2005). Thus, the genotyped RIL population will serve as an effective study system for characterizing a variety of agriculturally important traits, such as yield, grain quality and nutrition, stress tolerance, and water and nutrient use efficiency (Zhang 2007).
References
Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles ER, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Bennewitz J, Reinsch N, Kalm E (2002) Improved confidence intervals in quantitative trait loci mapping by permutation bootstrapping. Genetics 160:1673–1686
Broman KW, Wu H, Sen S, Churchill GA (2003) R/qtl: QTL mapping in experimental crosses. Bioinformatics 19:889–890
Da Y, Vanraden PM, Schook LB (2000) Detection and parameter estimation for quantitative trait loci using regression models and multiple markers. Genet Sel Evol 32:357–381
Doebley J, Stec A, Hubbard L (1997) The evolution of apical dominance in maize. Nature 386:485–488
Fan C, Xing Y, Mao H, Lu T, Han B, Xu C, Li X, Zhang Q (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112:1164–1171
Frary A, Nesbitt TC, Grandillo S, Knaap E, Cong B, Liu J, Meller J, Elber R, Alpert KB, Tanksley SD (2000) fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289:85–88
Gautier M, Barcelona RR, Fritz S, Grohs C, Druet T, Boichard D, Eggen A, Meuwissen THE (2006) Fine mapping and physical characterization of two linked quantitative trait loci affecting milk fat yield in dairy cattle on BTA26. Genetics 172:425–436
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M, Glazebrook J, Sessions A, Oeller P, Varma H, Hadley D, Hutchison D, Martin C, Katagiri F, Lange BM, Moughamer T, Xia Y, Budworth P, Zhong J, Miguel T, Paszkowski U, Zhang S, Colbert M, Sun WL, Chen L, Cooper B, Park S, Wood TC, Mao L, Quail P, Wing R, Dean R, Yu Y, Zharkikh A, Shen R, Sahasrabudhe S, Thomas A, Cannings R, Gutin A, Pruss D, Reid J, Tavtigian S, Mitchell J, Eldredge G, Scholl T, Miller RM, Bhatnagar S, Adey N, Rubano T, Tusneem N, Robinson R, Feldhaus J, Macalma T, Oliphant A, Briggs S (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296:92–100
Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, Spelman R, Georges M, Snell R (2002) Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res 12:222–231
Harjes C, Rocheford TR, Bai L, Brutnell TP, Kandianis CB, Sowinski SG, Stapleton AE, Vallabhaneni R, Williams M, Wurtzel ET, Yan J, Buckler ES (2008) Natural genetic variation in Lycopene Epsilon Cyclase tapped for maize biofortification. Science 319:330–333
Harushima Y, Yano M, Shomura A, Sato M, Shimano T, Kuboki Y, Yamamoto T, Lin SY, Antonio BA, Parco A, Kajiya H, Huang N, Yamamoto K, Nagamura Y, Kurata N, Khush GS, Sasaki T (1998) A high-density rice genetic linkage map with 2275 markers using a single F2 population. Genetics 148:479–494
Hittalmani S, Huang N, Courtois B, Venuprasad R, Shashidhar HE, Zhuang JY, Zheng KL, Liu GF, Wang GC, Sidhu JS, Srivantaneeyakul S, Singh VP, Bagali PG, Prasanna HC, McLaren G, Khush GS (2003) Identification of QTL for growth- and grain yield-related traits in rice across nine locations of Asia. Theor Appl Genet 107:679–690
Huang X, Feng Q, Qian Q, Zhao Q, Wang L, Wang A, Guan J, Fan D, Weng Q, Huang T, Dong G, Sang T, Han B (2009) High-throughput genotyping by whole-genome resequencing. Genome Res 19:1068–1076
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Ishimaru K (2003) Identification of a locus increasing rice yield and physiological analysis of its function. Plant Physiol 133:1083–1090
Ishimaru K, Ono K, Kashiwagi T (2004) Identification of a new gene controlling plant height in rice using the candidate-gene strategy. Planta 218:388–395
Ishimaru K, Yano M, Aoki N, Ono K, Hirose Y, Lin SY, Monna L, Sasaki T, Ohsugi R (2001) Toward the mapping of physiological and agronomic characters on a rice function map: QTL analysis and comparison between QTLs and expressed sequence tags. Theor Appl Genet 102:793–800
Jiang GH, Xu CG, Li XH, He YQ (2004) Characterization of the genetic basis for yield and its component traits of rice revealed by doubled haploid population. Acta Genetica Sinica 31:63–72
Jin J, Huang W, Gao JP, Yang J, Shi M, Zhu MZ, Luo D, Lin HX (2008) Genetic control of rice plant architecture under domestication. Nat Genet 40:1365–1369
Kao CH, Zeng ZB, Teasdale RD (1999) Multiple interval mapping for quantitative trait loci. Genetics 152:1203–1216
Kashiwagi T, Ishimaru K (2004) Identification and functional analysis of a locus for improvement of lodging resistance in rice. Plant Physiol 134:676–683
Konishi S, Izawa T, Lin SY, Ebana K, Fukuta Y, Sasaki T, Yano M (2006) An SNP caused loss of seed shattering during rice domestication. Science 312:1392–1396
Lander ES, Schork NJ (1994) Genetic dissection of complex traits. Science 265:2037–2048
Lander ES, Green P, Abrahamson J, Barlow A, Daly MJ, Lincoln SE, Newberg LA (1987) MAPMAKER: an interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1:174–181
Li C, Zhou A, Sang T (2006) Rice domestication by reducing shattering. Science 311:1936–1939
Li ZF, Wan JM, Xia JF, Zhai HQ (2003) Mapping quantitative trait loci underlying appearance quality of rice grains (Oryza sativa L.). Acta Genetica Sinica 30:251–259
Lin HX, Ashikari M, Yamanouchi U, Sasaki T, Yano M (2002) Identification and characterization of a quantitative trait locus, Hd9, controlling heading date in rice. Breed Sci 52:35–41
Liu X, Zhang H, Li H, Li N, Zhang Y, Zhang Q, Wang S, Wang Q, Wang H (2008) Fine-mapping quantitative trait loci for body weight and abdominal fat traits: effects of marker density and sample size. Poult Sci 87:1314–1319
Mackay TFC (2001) The genetic architecture of quantitative traits. Annu Rev Genet 35:303–339
Mauricio R (2001) Mapping quantitative trait loci in plants: uses and caveats for evolutionary biology. Nat Rev Genet 2:370–381
Mei HW, Li ZK, Shu QY, Guo LB, Wang YP, Yu XQ, Ying CS, Luo LJ (2005) Gene actions of QTLs affecting several agronomic traits resolved in a recombinant inbred rice population and two backcross populations. Theor Appl Genet 110(4):649–659
Mei HW, Xu JL, Li ZK, Yu XQ, Guo LB, Wang YP, Ying CS, Luo LJ (2006) QTLs influencing panicle size detected in two reciprocal introgressive line (IL) populations in rice (Oryza sativa L.). Theor Appl Genet 112:648–656
Qi J, Qian Q, Bu Q, Li S, Chen Q, Sun J, Liang W, Zhou Y, Chu C, Li X, Ren F, Palme K, Zhao B, Chen J, Chen M, Li C (2008) Mutation of the rice Narrow leaf1 gene, which encodes a novel protein, affects vein patterning and polar auxin transport. Plant Physiol 147:1947–1959
Sasaki A, Ashikari M, Ueguchi-Tanaka M, Itoh H, Nishimura A, Swapan D, Ishiyama K, Saito T, Kobayashi M, Khush GS, Kitano H, Matsuoka M (2002) Green revolution: a mutant gibberellin-synthesis gene in rice. Nature 416:701–702
Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, Konishi S, Yano M (2008) Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet 40:1023–1028
Song XJ, Huang W, Shi M, Zhu MZ, Lin HX (2007) A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39:623–630
Sutter N, Bustamante CD, Chase K, Gray MM, Zhao K, Zhu L, Padhukasahasram B, Karlins E, Davis S, Jones PG, Quignon P, Johnson GS, Parker HG, Fretwell N, Mosher DS, Lawler DF, Satyaraj E, Nordborg M, Lark KG, Wayne RK, Ostrander EA (2007) A single IGF1 allele is a major determinant of small size in dogs. Science 316:112–115
Tadege M, Sheldon CC, Helliwell CA, Upadhyaya NM, Dennis ES, Peacock WJ (2003) Reciprocal control of flowering time by OsSOC1 in transgenic Arabidopsis and by FLC in transgenic rice. Plant Biotechnol J 1(5):361–369
Tanksley SD (1993) Mapping polygenes. Annu Rev Genet 27:205–233
Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, Archibald AL, Haley CS, Buys N, Tally M, Andersson G, Georges M, Andersson L (2003) A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425:832–836
Visscher PM, Thompson R, Haley CS (1996) Confidence intervals in QTL mapping by bootstrapping. Genetics 143:1013–1020
Wan XY, Wan JM, Jiang L, Wang JK, Zhai HQ (2006) QTL analysis for rice grain length and fine mapping of an identified QTL with stable and major effects. Theor Appl Genet 112:1258–1270
Wang S, Basten CJ, Zeng ZB (2007) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh
Xue W, Xing Y, Weng X, Zhao Y, Tang W, Wang L, Zhou H, Yu S, Xu C, Li X, Zhang Q (2008) Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40:761–767
Yamamoto T, Sasaki T, Yano M (1997) Genetic analysis of spreading stub using indica/japonica backcrossed progenies in rice. Breed Sci 47:141–144
Yan CJ, Liang GH, Chen F, Li X, Tang SZ, Yi CD, Tian S, Lu JF, Gu MH (2003) Mapping quantitative trait loci associated with rice grain shape based on an indica/japonica backcross population. Acta Genetica Sinica 30:711–716
Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, Fuse T, Baba T, Yamamoto K, Umehara Y, Nagamura Y, Sasaki T (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12:2473–2484
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296:79–92
Yu CY, Liu YQ, Jiang L, Wang CM, Zhai HQ, Wan JM (2005) QTLs mapping and genetic analysis of tiller angle in rice (Oryza sativa L.). Acta Genetica Sinica 32:948–954
Yu B, Lin Z, Li H, Li X, Li J, Wang Y, Zhang X, Zhu Z, Zhai W, Wang X, Xie D, Sun C (2007) TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J 52:891–898
Yue B, Xue WY, Luo LJ, Xing YZ (2006) QTL analysis for flag leaf characteristics and their relationships with yield and yield traits in rice. Acta Genetica Sinica 33:824–832
Zhang Q (2007) Strategies for developing Green Super Rice. Proc Natl Acad Sci USA 104:16402–16409
Acknowledgments
We thank Zhou TY, Zong G, Gu BG, Shi JP, Hu WH, and Hu J for assistance with field management, phenotypic evaluation, and sample preparation. This work was supported by the Ministry of Science and Technology of China (the China Rice Functional Genomics Programs, Grant # 2006AA10A102), the Chinese Academy of Sciences (Grant # KSCX2-YW-N-024), and the National Natural Science Foundation of China (Grant # 30821004) to BH.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Sasaki.
L. Wang and A. Wang contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wang, L., Wang, A., Huang, X. et al. Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor Appl Genet 122, 327–340 (2011). https://doi.org/10.1007/s00122-010-1449-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-010-1449-8