
Abstract
Single nucleotide polymorphisms (SNPs) are indispensable in such applications as association mapping and construction of high-density genetic maps. These applications usually require genotyping of thousands of SNPs in a large number of individuals. Although a number of SNP genotyping assays are available, most of them are designed for SNP genotyping in diploid individuals. Here, we demonstrate that the Illumina GoldenGate assay could be used for SNP genotyping of homozygous tetraploid and hexaploid wheat lines. Genotyping reactions could be carried out directly on genomic DNA without the necessity of preliminary PCR amplification. A total of 53 tetraploid and 38 hexaploid homozygous wheat lines were genotyped at 96 SNP loci. The genotyping error rate estimated after removal of low-quality data was 0 and 1% for tetraploid and hexaploid wheat, respectively. Developed SNP genotyping assays were shown to be useful for genotyping wheat cultivars. This study demonstrated that the GoldenGate assay is a very efficient tool for high-throughput genotyping of polyploid wheat, opening new possibilities for the analysis of genetic variation in wheat and dissection of genetic basis of complex traits using association mapping approach.
Similar content being viewed by others


Introduction
Single nucleotide polymorphism (SNP) is a ubiquitous type of genetic variation in eukaryotic genomes. SNPs are ideally suited for the construction of high-resolution genetic maps, investigation of population evolutionary history and discovery of marker–trait associations in association mapping experiments (International HapMap Consortium 2007; Zhao et al. 2007; Aranzana et al. 2005). In association mapping, a dense set of SNP markers covering the entire genome is needed for finding a casual mutation or an SNP that is in linkage disequilibrium with the casual mutation (Aranzana et al. 2005; Flint-Garcia et al. 2005). Inclusion of a large number of individuals in an association mapping study is critical to have sufficient power of association test (Aranzana et al. 2005; Flint-Garcia et al. 2005). Construction of high-density genetic maps requires genotyping large progeny at a large number of loci (Troggio et al. 2007; International HapMap Consortium 2007). These applications, therefore, require genotyping platforms capable of producing multi-locus genotypes in a large panel of individuals. A number of genotyping technologies are currently available that satisfy these requirements. The recently developed genotyping platform Illumina BeadArray combined with the GoldenGate assay is able to genotype up to 1,536 polymorphic sites in 384 individuals in a single reaction (Oliphant et al. 2002). Molecular Inversion Probe (MIP) technology offers 12,000-plex SNP genotyping assay for genotyping of a limited number of individuals (Hardenbol et al. 2005). An open-source, medium throughput SNP genotyping platform has recently been developed for genotyping thousands of individuals at tens to hundreds of SNP loci (Macdonald et al. 2005). All these technologies are based on oligo ligation assay (OLA) (Oliphant et al. 2002).
The Illumina GoldenGate assay utilizes two allele-specific oligonucleotides (ASOs) to discriminate between the allelic states at an SNP locus (Fan et al. 2003). The discriminating nucleotides are located at the 3′ ends of the pair of ASOs. The third primer, referred to as the locus specific oligonucleotide (LSO), anneals downstream of ASO. This primer contains an address sequence (IllumiCode) unique for each SNP site. After annealing, the ASO with a 3′-end base complementary to the base at the SNP site is extended by polymerase and ligated to the corresponding LSO forming a PCR template. The extension and ligation reaction is performed at multiple (up to 1,536) SNP loci simultaneously. The templates are then PCR amplified using a set of three primers common to all SNP sites. Two of them are labeled with either Cy3 or Cy5 fluorescent dye and each anneals to one of the two ASO sequences. The third primer anneals to a common sequence in the LSOs. PCR products are annealed to an array composed of beads carrying oligonucleotides complementary to the IllumiCode sequences in the LSOs that are used for recognition of each SNP site. The ratio of the intensity of Cy3 and Cy5 fluorescence is used to determine the allelic state at an SNP site. Equal intensity of Cy3/Cy5 signal (ratio 1:1) corresponds to the heterozygous state; 1:0 or 0:1 signal ratios correspond to one of the alternative homozygous states at an SNP site. The actual genotype calling is performed after clustering of fluorescence intensity values taking into account variation in data collected from individual beads and averaging and removing outliers. Up to three genotype clusters could be obtained for each SNP site in a diploid organism.
Most of the high-throughput SNP genotyping platforms and the SNP data analysis algorithms were developed for generating and analyzing diploid genotype data. Genotyping of polyploid organisms, mostly represented by plant species, is complicated by the presence of two or more gene copies in the nucleus. A frequently used strategy to overcome this complication is selective PCR amplification using primers specific to one or another copy of the duplicated locus (Caldwell et al. 2004). However, this approach could not be easily scaled up and used for developing high-throughput genotyping assays.
Recently, the Illumina GoldenGate genotyping assays, also referred to as oligonucleotide pool assays (OPAs), were designed for complex plant genomes such as soybean and barley (Hyten et al. 2008; Rostoks et al. 2006). In both OPAs, approximately 90% of the SNP assays produced high-quality genotype calls (Rostoks et al. 2006; Hyten et al. 2008). However, even though barley and soybean genomes contain significant proportions of duplicated genes (Blanc and Wolfe 2004), the level of gene duplication and homology of the duplicated copies is far less than those in recently evolved polyploids, such as wheat.
Wheat species form a classical polyploid series of diploid, tetraploid and hexaploid species that evolved as follows. Tetraploid wheat Triticum turgidum (genome formula AABB) evolved via hybridization of two diploid species closely related to T. urartu (genome formula AA) and Aegilops speltoides (genome formula SS, where S is similar to B) (Sarkar and Stebbins 1956; Dvorak and Zhang 1990; Dvorak et al. 1993). Hybridization of T. turgidum with diploid Ae. tauschii (genome formula DD) resulted in the evolution of hexaploid wheat, T. aestivum (genome formula AABBDD) (Kihara 1944; McFadden and Sears 1946). Polyploid wheat genotyping currently relies on RFLP, AFLP, SSR, DArT or genome-specific STS markers (Gill et al. 1991; Lotti et al. 2000; Song et al. 2005; Akbari et al. 2006). However, none of these markers is easily amenable to high-throughput genotyping. Here we demonstrate that the Illumina BeadArray platform and GoldenGate SNP assay provide a fast, reliable and cost-effective method for the large-scale acquisition of SNP genotype data in polyploid wheat. Along with the recently developed resource of nearly 2,000 wheat loci harboring SNPs (http://wheat.pw.usda.gov/SNP/new/index.shtml) and advances in sequencing technologies making high-throughput SNP discovery a feasible task (Barbazuk et al. 2007; Margulies et al. 2005; Shendure et al. 2005), the Illumina BeadArray platform opens new possibilities for the genome-wide analysis of genetic variation in wheat.
Materials and methods
Plant material
A total of 91 polyploid wheat lines including 53 lines of wild tetraploid emmer wheat Triticum turgidum ssp. dicoccoides (henceforth T. dicoccoides), 29 lines of hexaploid T. aestivum (16 varieties and 13 landraces) and 9 lines of synthetic wheat derived from the hybridization of various tetraploid wheats with Ae. tauschii were genotyped at 96 SNP loci. The identity of these lines is summarized in supplementary Table S1. Plants were grown in a greenhouse and DNA was extracted from leaves of 3–4-week-old seedlings according to a described protocol (Dvorak et al. 1988).
Genotyping and data analysis
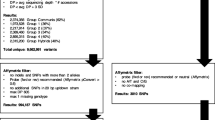
A set of 135 SNPs discovered in a panel of 32 lines of tetraploid and hexaploid wheat by sequencing of 92 gene fragments was downloaded from the Wheat SNP Database (http://wheat.pw.usda.gov/SNP/new/index.shtml). The total length of sequenced regions was 51,493 bp. The discovery panel included 10 accessions of wild emmer, 13 accessions of hexaploid wheat represented by landraces and 9 accessions of synthetic wheats (http://wheat.pw.usda.gov/SNP/new/index.shtml and Supplementary Table S1). The list of SNPs is provided in the Supplementary materials. Repetitive elements in the sequences detected by comparing them with the TREP (http://wheat.pw.usda.gov/ITMI/Repeats/) and GIRI (www.girinst.org) databases were masked. The SNP-harboring sequences were then submitted to Illumina for processing by Illumina® Assay Design Tool (ADT). ADT generates scores for each SNP that could vary from 0 to 1; SNPs with the scores above 0.6 have a high probability to be converted into a successful genotyping assay. In a set of 135 submitted SNPs, the ADT score varied from 0.18 to 0.99 with mean 0.85 (Table S2). A total of 96 SNP sites that were present at the frequency above 2 in the discovery panel and having ADT scores above 0.6 were selected for OPA design (Tables S2, S3). Out of 96 SNPs, 26 were in the wheat D-genome and 70 SNPs were in the A-genome.
A total of 150 ng of genomic DNA per plant was used for Illumina SNP genotyping at the UC Davis Genome center (www.genomecenter.ucdavis.edu/dna_technologies) using the Illumina BeadArray platform and GoldenGate Assay following the manufacturer’s protocol. The fluorescence images of an array matrix carrying Cy3- and Cy5-labeled beads were generated with the two-channel scanner. Raw hybridization intensity data processing, clustering and genotype calling were performed using the genotyping module in the BeadStudio package (Illumina, San Diego, CA, USA). Illumina developed a self-normalization algorithm that relies on information contained in each array. This algorithm adjusts for channel-dependent intensity variations, differences in the background between the channels, and possible crosstalk between the dyes. The normalization procedure implemented in the BeadStudio genotyping module includes outlier removal and background correction and scaling (details of this proprietary normalization algorithm could be obtained from Illumina, San Diego, CA). Before genotype calling, the trimmed mean intensities were calculated from the normalized intensity values obtained for each bead type on the array by rejecting outliers to ensure high quality of genotype data. Genotype calls were generated using the GenCall software incorporated into the BeadStudio package. This algorithm uses a Bayesian model to assign normalized intensity values to one of the three possible homozygous and heterozygous genotype clusters. In the presence of only two homozygous clusters, GenCall computes the location of a missing heterozygous cluster by simulating data using the artificial neural network (Shen et al. 2005). Since only two clusters were expected for homozygous polyploid wheat lines (see “Discussion” for details), the genotype clusters generated for each SNP locus by GenCall were edited manually after visual inspection of Cy3 and Cy5 fluorescence intensity clustering on two-dimensional Cartesian plots. SNPs that failed to show two-group clustering were excluded from the analysis.
Genotyping error rate was assessed by comparing SNP genotypes determined with the GoldenGate assay with those determined by Sanger sequencing. Trace files for 56 SNP-harboring gene loci were downloaded from the Wheat SNP project database (http://wheat.pw.usda.gov/SNP/new/index.shtml). Base calling and sequence assembly were performed using the phred/phrap and consed programs (Ewing and Green 1998; Ewing et al. 1998; Gordon et al. 1998). SNP discovery was performed with the polyphred program using the default settings (Stephens et al. 2006) followed by visual inspection of sequence trace files and manual verification of each discovered SNP. Genetic diversity, defined as the probability that two randomly chosen alleles from the population are different (Weir 1996, p. 150, 151), was calculated using the PowerMarker program (Liu and Muse 2005).
Results
Genotyping with the Illumina GoldenGate assay
The GoldenGate assay ASOs and LSOs may anneal to targets in two or three genomes in wheat, depending on ploidy. GoldenGate genotyping reaction performed on polyploid wheat genomic DNA is therefore expected to produce Cy3/Cy5 fluorescence ratios that differ from those expected for a diploid. Consider the origin of a new haplotype in the A-genome of tetraploid wheat (Fig. 1a, top panel). Mutation (A>T) in the A-genome creates a derived T base and an A/T SNP. In the B-genome, the ancestral A base remains unchanged. Hence, the SNP results in two homozygous genotypes, AAAA and TTAA (Fig. 1a). The ratio of A:T bases in these two genotypes are 1:0 and 1:1 (Fig. 1b). Since Cy3/Cy5 labeled ASOs anneal to the targets in both A- and B-genomes, the Cy3/Cy5 ratios will also be 1:0 and 1:1 (Fig. 1b). Following a similar reasoning, the genotypes will be AAAAAA and TTAAAA in hexaploid wheat, resulting in the 1:0 and 1:2 Cy3/Cy5 ratios (Fig. 1b). In both examples, the presence of the target sequence on homeologous chromosomes shifts the Cy3/Cy5 fluorescence ratio toward that of the ancestral allele of the SNP and affects genotype clustering. The SNP genotyping of heterozygous tetraploid and hexaploid plants would produce 3:1 and 5:1 Cy3/Cy5 ratios, respectively (Fig. 1b).

GoldenGate SNP genotyping in polyploid wheat. a Principles of GoldenGate SNP genotyping in tetraploid wheat. The top panel shows two genotypes of T. dicoccoides polymorphic for A/T SNP in the A-genome and monomorphic for the T base at the orthologous site in the B-genome. The bottom panel shows the same two genotypes except for the LSO annealing site in the B-genome showing a secondary mutation resulting in imperfect match. Upstream ASOs are blue and red and downstream LSO is black. b The expected Cy3/Cy5 fluorescence ratios for homozygous (AA, TT) and heterozygous (AT) diploid, tetraploid and hexaploid plants. The genotypes of the polymorphic sites are shown in parentheses
A 96-plex OPA included 70 A-genome SNPs and 26 D-genome SNPs that were discovered by sequencing gene fragments in the SNP discovery panel consisting of 32 lines of tetraploid and hexaploid wheat. For an SNP to be selected for this study, the minor allele count had to be ≥2 (6.3%) in the screening population. A total of 91 polyploid wheat lines including 53 lines of T. dicoccoides, 29 lines of hexaploid T. aestivum (16 cultivars and 13 landraces) and 9 lines of synthetic wheat were genotyped using GoldenGate assay. Clustering of Cy3 and Cy5 normalized intensities in a Cartesian plot was used to infer the SNP genotypes. A total of 8 (12%) SNPs in tetraploid wheat and 15 (15.6%) in hexaploid wheat that failed to generate the two-group clustering expected for homozygous lines were excluded from further analyses (Fig. 2). Genotype calls for the remaining 62 A-genome SNPs in tetraploid wheat and 81 A- and B-genome SNPs in hexaploid wheat could be grouped into one or two clusters depending on whether an SNP site was monomorphic or polymorphic (Fig. 3a, b). In the panel of hexaploid wheat lines, 8 SNP sites were monomorphic (all in the A-genome) and 73 were polymorphic (52 SNPs in the A-genome and 21 SNPs in the D-genome). In tetraploid wheat, 2 SNP sites were monomorphic and 60 SNP sites were polymorphic. As expected, there were more monomorphic SNP sites in cultivated wheat (25/81) than in the panel of 13 wheat landraces (9/81).

SNP sites failing to produce a two-cluster pattern. Genotyping plots were generated by graphing normalized Cy3 and Cy5 fluorescence intensities

Genotype calls in a sample of 53 accessions of tetraploid T. dicoccoides (a) and 38 accessions of hexaploid T. aestivum (b). The accession numbers of mapped EST unigenes are shown on the top of each panel. Genotyping plots were generated by plotting Cy3 and Cy5 normalized fluorescence intensities. a SNP sites polymorphic in the A-genome of T. dicoccoides. b SNP sites that were polymorphic in the hexaploid wheat A- or D-genomes
Concordance between the SNP genotypes determined with the GoldenGate assay and those determined with a standard Sanger sequencing approach was used to assess the accuracy of genotyping (Table 1). These comparisons were made for 56 SNP sites in 10 accessions of T. dicoccoides and 22 accessions of hexaploid wheat, although not all accessions could be compared for some SNPs because of low-quality sequence reads (Table 1). Out of 1,304 SNP genotype calls generated by GoldenGate assay (403 SNP genotypes in tetraploid wheat and 901 SNP genotypes in hexaploid wheat) 1,292 were identical with the genotypes obtained by Sanger sequencing. This results in genotyping accuracy rate of 100% in tetraploid wheat (data not shown) and 99% in hexaploid wheat (Table 1).
Clustering of Illumina genotype calls may fail in an allopolyploid for the same reasons as in a diploid and also for allopolyploidy-specific reasons. Oligonucleotide probes designed for SNP detection (ASO and LSO) anneal to short regions flanking the query SNPs. Since there are three nearly identical homeologous sets of chromosomes present in the polyploid wheat genome, genotyping oligos have up to three homeologous targets to bind to. If sequences of these targets are identical, genotyping oligos will bind with equal efficiency to each of the homeologs. However, divergence of the annealing sites in homeologous chromosomes could change oligonucleotide binding efficiency and interfere with genotyping assay resulting in its failure (Fig. 1a, bottom panel). To assess if the divergence of homeologous sequences at the genotyping oligonucleotide binding sites could be the cause of genotyping assay failures or inadequate clustering, regions flanking the two SNPs (wsnpBE443187_5A_2 and wsnpBE489901_2D_3) excluded from the analyses were investigated (Fig. 2). For both SNPs sequences from homeologous chromosomes were available. Sequences flanking the SNP site in the wsnpBE443187_5A_2 locus, which is located in the A-genome were compared with the orthologous sequences from both the B- and D-genomes of polyploid wheat. Similarly, sequences flanking the SNP site in the wsnpBE489901_2D_3 locus, which is located in the D-genome were compared with the B-genome orthologous sequence. In both cases, divergence found at the oligonucleotide annealing sites could be associated with the failure to generate defined clusters on the Cartesian plots (Fig. 2). At the wsnpBE443187_5A_2 SNP site, both ASOs and LSO A-genome annealing sites differed by one nucleotide either in the B- or D-genome. At the wsnpBE489901_2D_3 SNP site, the A-genome ASOs annealing site differed from that in the B-genome by a dinucleotide indel.
Diversity estimation with the Illumina GoldenGate assay
Genetic diversity of the A- and D-genomes of hexaploid wheat was 0.31 and 0.34, respectively. Diverse panel of 13 wheat landraces demonstrated a higher level of genetic diversity (A-genome −0.32; D-genome −0.35) than the panel of 16 wheat cultivars (A-genome −0.23; D-genome −0.31). The observed 28 and 11% reduction in the levels of genetic diversity in the A- and D-genomes, respectively, most likely results from the bottleneck caused by selection of only limited number of accessions for the development of adapted cultivars. Genetic diversity of the A-genome of tetraploid wild emmer (0.25) was very close to that of the A-genome of cultivated wheat (0.23). However, when only shared set of SNP loci (55) was compared, the level of genetic diversity in the A-genome of polyploid wheat (0.3) was higher than that in the A-genome of tetraploid wild emmer (0.26). It should be noted, however, that provided estimates of diversity need to be treated cautiously. Since only common SNP variants were used for genotyping assay design, obtained diversity statistic is different from the one that can be obtained using all SNPs. This type of bias, known also as an ascertainment bias, is hard to control and should be avoided if the purpose of study is the inference of population history (Clark et al. 2005; Brumfield et al. 2003).
Mean genetic diversity was also computed for a subset of data for which both genotype calls and nucleotide sequences were available. Mean genetic diversity was 0.33 and 0.36 in the A- and D-genomes of T. aestivum, respectively, and 0.26 in the A-genome of T. dicoccoides. This simple descriptive statistic obtained by Illumina GoldenGate genotyping was compared with that calculated for all SNPs detected by sequencing (http://wheat.pw.usda.gov/SNP/new/index.shtml) (Fig. 4). Estimates of genetic diversity based on nucleotide sequences were 0.26 and 0.23 for the A- and D-genomes of T. aestivum, respectively, and 0.18 for the A-genome of T. dicoccoides. Mean genetic diversity calculated using SNPs derived from DNA sequences was lower than that calculated using SNPs obtained with the Illumina GoldenGate assay. The observed discrepancy between the Illumina GoldenGate data and nucleotide sequence data could be attributed to a bias introduced during selection of SNPs for the Illumina OPA design. Only those SNPs that present at frequency 2 and higher in the discovery population were selected for assay design. It could be clearly seen in Fig. 4 that the frequency spectrum of SNPs in OPA is shifted toward the high-frequency variants compared to that in Sanger sequence data.

Distribution of minor allele frequencies of SNPs in the A- and D-genomes of T. aestivum and the A-genome of T. dicoccoides detected with the GoldenGate assay and sequencing in the SNP discovery panel
Discussion
All analyses of SNP genotyping results in this study assumed that the genotypes have been obtained from homozygous plants. Due to high rates of self-pollination, this assumption holds for most of wheat landraces, wheat breeding lines and plants collected from wild populations. Genotyping of a mixture of homozygous and heterozygous plants, a situation prevailing in mapping populations generated from bi-parental crosses, would probably be more challenging due to the tight clustering of fluorescence intensity ratio values on genotyping plots. Apparently, it will be problematic to generate reliable genotype calls from such data. This limitation needs to be considered if the Illumina GoldenGate assay is planned to be used in gene mapping projects using bi-parental cross populations and can be overcome by using homozygous plants produced either by several generations of self-pollination or doubled-haploid technique.
The proportion of SNPs that could be converted into working assays is a very important factor that influences the cost of genotyping assay development. The presence of repetitive elements, palindromes and neighboring polymorphisms around a query SNP is known to affect the conversion rate (Fan et al. 2003). Homology search using two databases of repetitive elements was performed here during the selection of SNPs for OPA design to increase this rate. All sequences showing similarity to known repeats were masked before submitting them to Illumina for assay design. In addition, Illumina used its own proprietary bioinformatical screening procedures implemented in ADT to increase the assay conversion rate.
In polyploid species devoid of genome sequence data, such as wheat, the task of SNP selection with high “designability scores” is additionally complicated by the presence of multiple targets within the nucleus. Since GoldenGate assay relies on annealing of genotyping oligonucleotides to regions flanking a query SNP, the divergence of orthologous sequences in homeologous chromosomes or undetected polymorphisms could interfere with annealing of genotyping oligonucleotides and genotyping reactions. For example, the nucleotide sequence of the B-genome annealing sites might have diverged from the A-genome sequence and harbor mutations (Fig. 1a, bottom panel). Such mutations will reduce oligonucleotide annealing efficiency with the B-genome target, which will negatively affect clustering and reduce genotype calling rate. Interference of secondary SNPs at oligonucleotide annealing sites with genotyping accuracy has been demonstrated in Drosophila (Macdonald et al. 2005). In the present study, poor genotype call clustering (more than two clusters) was encountered at 8 SNP sites in tetraploid wheat (12%) and 15 sites in hexaploid wheat (15.6%). Homology of the orthologous annealing sequences was investigated at two of these sites. Divergent mutations were encountered at both sites suggesting that this may be an important source of clustering failure in genotyping of polyploids. To minimize the effect of divergent mutations on performance of genotyping assays, all copies of orthologous sequences in the wheat nucleus should be analyzed. However, this analysis could be performed only for a limited number of SNP loci until a complete wheat genome sequence is produced.
The presence of paralogous genes within genome can also have negative impact on genotyping. High incidence (27.7%) of recently duplicated genes in the wheat genome (Akhunov et al. 2003) increases the chance of designing genotyping assays targeting paralogous duplications. This could be avoided by additional analysis of genomic regions prior to genotyping assay design. Paralogs can be identified by sequence analysis of PCR amplicons or Southern hybridization analysis. For example, wheat EST database is an excellent resource for selecting non-repetitive targets in the wheat genome (http://wheat.pw.usda.gov/wEST/).
Despite the complexities intrinsic to polyploidy and large sizes of the wheat genomes, a high level of genotyping accuracy, 99–100% after removal of SNPs failed to produce two clusters on Cartesian plots, was achieved. This accuracy level is comparable to that achieved with the Illumina GoldenGate assays in human populations (99.7–99.9%) after applying quality cutoffs (Fan et al. 2003; Steemers and Gunderson 2007). A conversion rate of 89 and 84% achieved in tetraploid and hexaploid wheat, respectively, for SNPs selected from the wheat SNP database was also comparable to a conversion rate for human SNPs. Only SNPs that had a minor allele frequency of 6.3% or higher (two or more hits in the screening population) were used for wheat OPA design. Fan et al. (2003) reported a conversion rate of 81% for randomly selected SNPs from dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) and 94–96% for “double-hit” SNPs (SNPs encountered in database two or more times). Enhanced conversion rates for double hit SNPs in both studies argues for selection of SNPs based on their frequency in the screening population to minimize conversion failures. This procedure also increases the chance that the selected SNP will be polymorphic in genotyped populations.
Another potential source of genotyping errors in wheat is the presence of identical polymorphisms at orthologous loci in homeologous chromosomes (shared polymorphisms). Shared polymorphisms could be either inherited from the diploid ancestors or, less likely, evolved independently in polyploid wheat. The presence of a shared polymorphism A/T in the two genomes of tetraploid wheat would result in four possible genotypes: AAAA, TTAA, AATT and TTTT. The TTAA and AATT genotypes would likely appear identical in the Cy3/Cy5 plot. Three clusters would therefore be expected. If the TTAA and AATT genotypes would not appear identical in the plot, four clusters would be possible. To assess the significance of shared polymorphism for genotyping in polyploid wheat, 82 inter-genomic sequence alignments (total length of aligned sequence is 35,715 bp; data not shown) were compared. No shared polymorphism was found, indicating that shared polymorphisms are of negligible significance for Illumina genotyping in polyploid wheat.
A source of genotyping errors that was not considered in our study is the presence of SNP sites with three or four alleles in polypoid wheat. These SNP sites are inherently very rare in populations because their origin requires two or more independent mutations at the same site. The probability of such mutations is extremely low given spontaneous mutation rates of 10−8–10−9 mutations at site−1 year−1 in eukaryotic genomes. We therefore expect that this class of SNPs has a negligible effect on genotyping error rate in wheat.
Our study calls for caution in using a limited number of SNPs, particularly if preselected on the basis of their polymorphism and allele frequencies in the screening population, in making inferences about diversity and evolutionary history of populations. First of all, since only polymorphic sites are investigated, the estimates of genetic diversity are expected to be biased upward. For instance, genetic diversity of the wheat D-genome estimated in this study using OPA was higher than the genetic diversity of the wheat A-genome. Genetic diversity based on the Sanger sequence data showed that the A-genome is more diverse than the D-genome (see “Results”). A survey of a larger number of gene loci by sequencing also demonstrated that the genetic diversity of the wheat D-genome is significantly lower than genetic diversity of the wheat A-genome (unpublished data). The observed discrepancy results from the bias introduced by the selection of high-frequency SNPs for OPA design (Fig. 4). Preferential use of SNPs that are present at intermediate frequencies in a population will affect the population statistics relying on the frequency spectrum of SNPs, such as genetic diversity, population differentiation and linkage disequilibrium (Clark et al. 2005; Brumfield et al. 2003). For example, medium frequency SNPs will most likely be shared between populations and, therefore, the extent of population differentiation will be underestimated. It has also been suggested that focusing on common SNPs might have an effect on the power of detecting association between genetic variants and casual mutations. The deficit of rare SNPs could result in reduced power of association analysis when causal variants are rare in a population (Clark et al. 2005).
The distribution of SNPs in cultivated wheat gene pool defines their utility for breeding and marker-assisted selection applications. Our results suggest that the panel of wheat lines used for SNP discovery captured significant proportion of genetic variation present in modern cultivated wheat. About 70% of SNPs preselected on the basis of their polymorphisms in the discovery panel were also polymorphic in a set of 16 wheat cultivars (Table S1). This is consistent with the conclusion based on the analysis of 359 SNPs derived from the same database (Chao et al. 2009). Selection of SNPs with the frequency above 2 in the discovery panel might have also increased the probability of finding polymorphic SNP sites in the cultivated gene pool. High-frequency SNPs had a higher chance to pass through the population bottleneck that apparently accompanied the development of adapted cultivars. This is evident from the comparison of the genetic diversity of landraces and cultivars. The latter lost 28 and 11% of their genetic diversity in the A- and D-genomes, respectively, compared to that in landraces (see “Results”). Loss of genetic diversity in wheat varieties compared to that in the populations of landraces has also been reported by Chao et al. (2009).
Conclusion
It is demonstrated that SNPs can be genotyped in polyploid wheat pure lines with the Illumina BeadArray technology coupled with GoldenGate assay without the need of a prior PCR amplification step. About 89 and 84% of SNPs in tetraploid and hexaploid wheat, respectively, could be converted into successful genotyping assays. After eliminating SNPs showing low-quality clustering, the accuracy of genotyping was 100 and 99% for tetraploid and hexaploid wheat, respectively. The utility of SNPs selected from the wheat SNP database for genotyping of wheat varieties was demonstrated. The Illumina BeadArray platform, therefore, can offer a cost- and time-effective approach to genotyping of thousands of polyploid wheat lines at thousands of polymorphic sites. This technology represents an excellent tool for studying genetic architecture of complex traits, association mapping and, with proper safeguards, evolutionary forces that shaped the genetic diversity of polyploid wheat and its wild relatives.
References
Akbari M, Wenzl P, Caig V, Carling J, Xia L, Yang SY, Uszynski G, Mohler V, Lehmensiek A, Kuchel H, Hayden MJ, Howes N, Sharp P, Vaughan P, Rathmell B, Huttner E, Kilian A (2006) Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor Appl Genet 113:1409–1420
Akhunov ED, Goodyear AW, Geng S, Qi LL, Echalier B, Gill BS, Miftahudin Gustafson JP, Lazo G, Chao SM, Anderson OD, Linkiewicz AM, Dubcovsky J, La Rota M, Sorrells ME, Zhang DS, Nguyen HT, Kalavacharla V, Hossain K, Kianian SF, Peng JH, Lapitan NLV, Gonzalez-Hernandeiz JL, Anderson JA, Choi DW, Close TJ, Dilbirligi M, Gill KS, Walker-Simmons MK, Steber C, McGuire PE, Qualset CO, Dvorak J (2003) The organization and rate of evolution of wheat genomes are correlated with recombination rates along chromosome arms. Genome Res 13:753–763
Aranzana MJ, Kim S, Zhao K, Bakker E, Horton M, Jakob K, Lister C, Molitor J, Shindo C, Tang C, Toomajian C, Traw B, Zheng H, Bergelson J, Dean C, Marjoram P, Nordborg M (2005) Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet 1:e60
Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS (2007) SNP discovery via 454 transcriptome sequencing. Plant J 51:910–918
Blanc G, Wolfe KH (2004) Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 16:1667–1678
Brumfield RT, Beerli P, Nickerson DA, Edwards SV (2003) The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol Evol 18:249–256
Caldwell KS, Dvorak J, Lagudah ES, Akhunov E, Luo MC, Wolters P, Powell W (2004) Sequence polymorphism in polyploid wheat and their D-genome diploid ancestor. Genetics 167:941–947
Chao S, Zhang W, Akhunov E, Sherman J, Ma Y, Luo MC, Dubcovsky J (2009) Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.) cultivars. Mol Breed 23:23–33
Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R (2005) Ascertainment bias in studies of human genome-wide polymorphism. Genome Res 15:1496–1502
Dvorak J, Zhang HB (1990) Variation in repeated nucleotide sequences sheds light on the phylogeny of the wheat B and G genomes. Proc Natl Acad Sci USA 87:9640–9644
Dvorak J, McGuire PE, Cassidy B (1988) Apparent sources of the A genomes of wheats inferred from the polymorphism in abundance and restriction fragment length of repeated nucleotide sequences. Genome 30:680–689
Dvorak J, di Terlizzi P, Zhang HB, Resta P (1993) The evolution of polyploid wheats: identification of the A genome donor species. Genome 36:21–31
Ewing B, Green P (1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res 8:186–194
Ewing B, Hillier L, Wendl M, Green P (1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res 8:175–185
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L, Hunt S, McBride C, Bibikova M, Rubano T, Chen J, Wickham E, Doucet D, Chang W, Campbell D, Zhang B, Kruglyak S, Bentley D, Haas J, Rigault P, Zhou L, Stuelpnagel J, Chee MS (2003) Highly parallel SNP genotyping. Cold Spring Harbor Symp Quant Biol 68:69–78
Flint-Garcia SA, Thuillet AC, Yu JM, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for QTL dissection. Plant J 44:1054–1064
Gill KS, Lubbers EL, Gill BS, Raupp WJ, Cox TS (1991) A genetic linkage map of Triticum tauschii (DD) and its relationship to the D genome of bread wheat (AABBDD). Genome 34:362–374
Gordon D, Abajian C, Green P (1998) Consed: a graphical tool for sequence finishing. Genome Res 8:195–202
Hardenbol P, Yu F, Belmont J, MacKenzie J, Bruckner C, Brundage T, Boudreau A, Chow S, Eberle J, Erbilgin A, Falkowski M, Fitzgerald R, Ghose S, Iartchouk O, Jain M, Karlin-Neumann G, Lu X, Miao X, Moore B, Moorhead M, Namsaraev E, Pasternak S, Prakash E, Tran K, Wang Z, Jones HB, Davis RW, Willis TD, Gibbs RA (2005) Highly multiplexed molecular inversion probe genotyping: over 10, 000 targeted SNPs genotyped in a single tube assay. Genome Res 15:269–275
Hyten DL, Song Q, Choi IY, Yoon MS, Cregan PB (2008) High-throughput genotyping with the GoldenGate assay in the complex genome of soybean. Theor Appl Genet 116:945–952
International HapMap Consortium (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861
Kihara H (1944) Discovery of the DD-analyser, one of the ancestors of Triticum vulgare (in Japanese). Agric Hortic 19:13–14
Liu K, Muse SV (2005) PowerMarker: integrated analysis environment for genetic marker data. Bioinformatics 21:2128–2129
Lotti C, Salvi S, Pasqualone A, Tuberosa R, Blanco A (2000) Integration of AFLP markers into an RFLP-based map of durum wheat. Plant Breed 119:393–401
Macdonald SJ, Pastinen T, Genissel A, Cornforth TW, Long AD (2005) A low-cost open-source SNP genotyping platform for association mapping applications. Genome Biol 6:R105
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer MLI, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
McFadden ES, Sears ER (1946) The origin of Triticum spelta and its free-threshing hexaploid relatives. J Hered 37:81–89
Oliphant A, Barker DL, Stuelpnagel JR, Chee MS (2002) BeadArray technology: enabling an accurate, cost-effective approach to high-throughput genotyping. Biotechniques Suppl 5:6–58
Rostoks N, Ramsay L, Mackenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF, Graner A, Close TJ, Waugh R (2006) Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA 103:18656–18661
Sarkar P, Stebbins GL (1956) Morphological evidence concerning the B genome in wheat. Am J Bot 43:297–304
Shen R, Fan JB, Campbell D, Chang W, Chen J, Doucet D, Yeakley J, Bibikova M, Wickham Garcia E, McBride C, Steemers F, Garcia F, Kermani BG, Gunderson K, Oliphant A (2005) High-throughput SNP genotyping on universal bead arrays. Mutat Res 573:70–82
Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM (2005) Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309:1728–1732
Song Q-J, Shi J-R, Singh S, Fickus EW, Costa JM, Lewis J, Gill BS, Ward R, Cregan PB (2005) Development and mapping of microsatellite (SSR) markers in wheat. Theor Appl Genet 110:550–560
Steemers FJ, Gunderson KL (2007) Whole genome genotyping technologies on the BeadArray platform. Biotechnol J 2:41–49
Stephens M, Sloan JS, Robertson PD, Scheet P, Nickerson DA (2006) Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat Genet 38:375–381
Troggio M, Malacarne G, Coppola G, Segala C, Cartwright DA, Pindo M, Stefanini M, Mank R, Moroldo M, Morgante M, Grando MS, Velasco R (2007) A dense single-nucleotide polymorphism-based genetic linkage map of grapevine (Vitis vinifera L.) anchoring Pinot Noir bacterial artificial chromosome contigs. Genetics 176:2637–2650
Weir BS (1996) Genetic data analysis II. Sinauer Associates Inc., Sunderland
Zhao K, Aranzana MJ, Kim S, Lister C, Shindo C, Tang C, Toomajian C, Zheng H, Dean C, Marjoram P, Nordborg M (2007) An Arabidopsis example of association mapping in structured samples. PLoS Genet 3:e4
Acknowledgments
We would like to thank Vanessa Rashbrook for the help with Illumina GoldenGate SNP genotyping and the four anonymous reviewers for helpful comments on the previous versions of the manuscript. The work has been supported by National Science Foundation Plant Genome Research Program award no. DBI-0321757.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Schulman.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Akhunov, E., Nicolet, C. & Dvorak, J. Single nucleotide polymorphism genotyping in polyploid wheat with the Illumina GoldenGate assay. Theor Appl Genet 119, 507–517 (2009). https://doi.org/10.1007/s00122-009-1059-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-009-1059-5