
Abstract
Type 1 diabetes (T1D) is one of the most studied archetypal organ-specific autoimmune diseases. Although many clinical, epidemiological, and pathological characteristics have been described, there are still important issues which need to be resolved as these will have a major impact on the development of future antigen-specific immunotherapies. An important question relates to T lymphocytes in the development of the disease, in particular their role in the destruction of insulin-producing beta cells. Since the discovery that certain class II histocompatibility complex molecules (HLA) are linked to the development of T1D, much research has focused on CD4+ helper T lymphocytes; however, recent studies highlight class I HLA molecules as an independent risk factor; hence, research into the role played by CD8+ cytotoxic T lymphocytes has gained momentum. In this review, we summarize recent studies clarifying the role played by both sets of autoreactive T lymphocytes in T1D, discuss the targets recognized by these cells and their phenotype in T1D patients. Finally, we will examine the possible generation of regulatory CD8+ T lymphocytes upon different immuno-intervention strategies.
Similar content being viewed by others


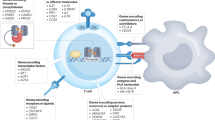
Introduction
Type 1 diabetes (T1D) is an organ-specific autoimmune disease that results from the destruction of the insulin-producing islet beta cells in the pancreas. Typically, the disease is more frequently diagnosed in children and adolescents, although a significant percentage of cases are reported amongst adults, and in some cases, even amongst some individuals originally classified as presenting with type 2 diabetes (T2D), suggesting disease heterogeneity in T1D (Arif et al. 2014; Atkinson 2012).
The incidence of T1D shows extensive global variation as shown by several studies: the EURODIAB ACE study group reported an annual incidence range of 3.2–40.2 cases per 100,000 per year in different European countries, an incidence that has doubled in the last 20 years, mainly in young children (EURODIAB ACE Study Group 2000). In the US, the SEARCH for diabetes in the Youth Consortium reported an increased incidence of the disease in a 9 year period (2001–2009) from 1.48 per 1000 up to 1.93 per 1000 in patients under 19 years of age (Dabelea et al. 2014), a 21.1% increase during the 9 years of the study. In 2015, the number of children with T1D exceeded half a million for the first time, with the highest incidence shown in European countries (International Diabetes Federation 2015). The reasons for this escalation are still unclear but may be due to changes in environmental risk factors, early events in the womb, diet early in life, or viral infections (International Diabetes Federation 2015).
Several discoveries in the early days of T1D research suggested that the disease was autoimmune in nature. The presence of islet autoantibodies targeting beta cells was first described in 1974 and since then many of the antigens targeted by these autoantibodies have been identified (Koo et al. 2014), including insulin, glutamic acid decarboxylase 65 (GAD65), the insulinoma-antigen 2 (IA-2), and more recently zinc transporter 8 (ZnT8). The presence of these autoantibodies is used in the clinic to confirm diagnosis, and they can be also used to predict the development of the disease in prediabetic individuals (Verge et al. 1996; Wenzlau and Hutton 2013).
The second important feature that points to the participation of the immune system in the etiology of T1D is the presence of an infiltration of the pancreatic islets by leukocytes, described as “insulitis”. Insulitis was first described in the mouse model of the disease, the non-obese diabetic (NOD) mouse, and although several cell types were found, T lymphocytes predominated (Anderson and Bluestone 2005). Insulitis in human T1D patients is difficult to evaluate partly due to the difficulty in obtaining pancreatic biopsies from recently diagnosed patients; however, when such samples were available, it was evident that insulitic infiltrations were indeed present in T1D subjects, and that the majority of the immune cells are lymphocytes.
CD8+ cytotoxic T lymphocytes were the more abundant T lymphocytes present in the insulitic infiltrations (Foulis et al. 1991; Itoh et al. 1993; Willcox et al. 2009). Moreover, class I human leukocyte antigen (HLA) is hyper-expressed in islet and endothelial cells in the majority of pancreatic biopsies from patients presenting with insulitis, and even in up to 20% of pancreatic biopsies from patients without insulitis (Itoh et al. 1993). These data suggests that the recognition of islet autoantigens presented by class I HLA molecules to cytotoxic T cells could be an important effector mechanism leading to the destruction of the beta cells.
Genetics: Focus on HLA
Multiple genome-wide scan studies for linkage with T1D have shown that T1D is a multigenic disease (Concannon et al. 2005, 2009), and these studies have consistently shown that the major histocompatibility complex (MHC) region, located in human chromosome 6, has the strongest linkage to T1D. Due to the strong linkage disequilibrium between the different loci present in the MHC region, it was originally difficult to identify the participation of different genes within this region. However, with increasing sample size and other methodological improvements it was possible to identify an independent effect for particular HLA alleles. Genetic studies have shown that particular combinations of class II HLA alleles of the DRB1, DQA1 and DQB1 loci determine the extent of risk of developing T1D. High genetic risk conferring susceptibility to T1D have been reported for the DR3-DQ2 (DRB1*0301-DQA1*0501-DQB1*0201) and DR4-DQ8 haplotypes (DRB1*0401-DQA1*0301-DQB1*0302), in particular for heterozygous subjects (Erlich et al. 2008). Since the physiological role of these class II molecules is to present peptide epitopes to CD4+ T lymphocytes, most studies focused on the role of CD4+ T lymphocytes in T1D pathogenesis.
Although class II molecules were the first to be shown to confer susceptibility to or protection from T1D more recent studies have shown that class I HLA molecules constitute an independent risk factor contributing to T1D development. Several molecules from class I HLA-A, -B and -C loci, such as the A*02:01, A*24:02, B*39:06, B*18:01, and C*05:01, have been identified as predisposing alleles. Furthermore, in the case of the A*02:01 allele, functional in vivo data indicates that this HLA molecule is able to select cytotoxic CD8+ T cells capable of destroying insulin-producing beta cells. Transgenic expression of A*02:01 in a mouse model with a NOD genetic background shows accelerated diabetes development as compared to non-transgenic littermates (Marron et al. 2002).
One hypothesis to explain the association of certain HLA molecules to the development of T1D is that these molecules present particular peptide epitopes from autoantigens targeted in the disease, such as preproinsulin or GAD65, whereas those HLA molecules that confer protection present different peptide epitopes that may elicit regulatory responses.
T Lymphocytes in T1D: Targets and Peripheral Detection
As described above, the insulitic infiltrates are mainly comprised of both CD4+ and CD8+ lymphocytes, with a predominance of cytotoxic T cells (Hanafusa and Imagawa 2008; Willcox et al. 2009). The higher representation of cytotoxic T lymphocytes among the inflammatory infiltrates is probably due to the fact that these cells might be the main effectors responsible for the destruction of the insulin-producing beta cells. Support for this idea comes from recent data which showed that insulin-specific cytotoxic T cell clones were capable of specifically destroying beta cells from human islets in vitro (Skowera et al. 2008); moreover, high levels of glucose accelerated this destruction, possibly due to a higher presentation of insulin peptide epitopes which activated the infiltrating cytotoxic T cells. Despite this dominance of T lymphocytes in the inflammatory infiltrates, other immune cells are also found, including B cells and macrophages. Furthermore, the composition of the cellular infiltrate is related to islet destruction and age of T1D onset (Arif et al. 2014; Leete et al. 2016).
Several critical questions arise regarding the T cells infiltrating the islets in T1D patients: (i) what are the targets recognized by the infiltrating autoreactive T lymphocytes? and (ii) is there any correlation between T cells present in the insulitic lesion and those detected in the peripheral blood?
The answer to the first question constitutes a research field that has been growing since the discovery of autoreactive T cells as the main mediators in the development of T1D. Several autoantigens have been identified, with three being the most prominent, probably due to the ease of detection of autoantibodies against them: pre-proinsulin, GAD65 and IA-2 (Di Lorenzo et al. 2007). Since the identification of these antigens many peptide epitopes derived from these and linked to the development of T1D have been identified, both in the NOD mouse model and in human patients (Babad et al. 2010; Di Lorenzo et al. 2007).
Besides these three main autoantigens, other targets of the autoimmune response have been identified (Han et al. 2013). For some of these novel autoantigens, there is data suggesting their significant role in the development of T1D, such as the existence of T-cell clones capable of transferring the disease or immunotherapy using antigenic peptides derived from these autoantigens (Table 1). These include islet-specific glusose-6-phosphatase catalytic subunit-related protein (IGRP), ZnT8 and, interestingly, the neurotropic factor S100beta and the glial fibrillary acidic protein (GFAP) (Gomez-Tourino et al. 2010, 2015; Lieberman et al. 2003; Trudeau et al. 2003; Winer et al. 2003), the latter two being antigens not expressed by the beta cells themselves but by a group of nerve cells surrounding the islets called the peri-islet Schwann cells (pSC) which makes their role in T1D pathogenesis controversial.
IGRP antigen was first identified as the antigen targeted by a prevalent population of pathogenic CD8+ T cells in NOD mice (Lieberman et al. 2003) and also by human T lymphocytes (Ouyang et al. 2006). Its relevance to T1D derives from the fact that in the NOD mice the number of IGRP-specific CD8+ T cells correlate with the future development of clinical disease (Trudeau et al. 2003).
ZnT8 antigen was first identified as a major autoantigen in human T1D in 2007 (Wenzlau et al. 2007); it has been reported that detection of ZnT8-specific autoantibodies enhance disease diagnosis, reducing the percentage of autoantibody-negative T1D patients. Furthermore, a significant percentage of single autoantibody-positive patients were in fact multiple antibody-positive after the inclusion of these antibodies in screening (Wenzlau et al. 2008).
The discovery that cells other than beta cells, such as the pSC cells, were also targeted by the autoimmune response in the islets of Langerhans led to the identification of two new autoantigens, the neurotropic factor S100beta and GFAP (Winer et al. 2003). Autoantibodies against these two antigens can be detected not only in the plasma of T1D patients (Gomez-Tourino et al. 2010; Winer et al. 2003) but also in up to a 30% of patients with T2D (Gomez-Tourino et al. 2010; Winer et al. 2011), where these autoantibodies appear to mediate the insulin-resistant state in these patients (Winer et al. 2011). However, the role played by autoantibodies in the development of insulin resistance and diabetes is still controversial and more studies are needed to resolve this. Both S100beta and GFAP also induce T cell responses both in the NOD mice and human T1D patients (Banwell et al. 2008; Gomez-Tourino et al. 2015; Serre et al. 2015; Standifer et al. 2006; Tsui et al. 2008; Winer et al. 2003): for S100beta, several peptide epitopes presented by HLA DR4 molecule were identified and T cell responses were detected in both T1D and T2D patients. Furthermore, pro-inflammatory responses characterized by the secretion of interferon (IFN)-γ were seen more frequently in T1D patients compared to healthy donors (Gomez-Tourino et al. 2015). Interestingly, responses seen against some of the epitopes were also found in long-standing T1D patients, indicating that these autoreactive T cells may contribute to the complications arising in these patients. Furthermore, there is a sustained response against S100beta during the course of the disease and several years after diagnosis. In the NOD mice, S100beta peptide epitopes recognized by CD4+ T lymphocytes have been described and, although presented by a completely different MHC molecule (the I-Ag7 molecule, homologous to the HLA-DQ8 in humans), the dominant peptide epitopes are similar to those presented by HLA-DR4 in human T1D as we previously reported (Serre et al. 2015).
In the case of the neuronal antigen GFAP, several peptide epitopes have been identified in NOD mice, targeted by both CD4+ and CD8+ T lymphocytes; interestingly, several of those epitopes are also recognized by human T cells (Tsui et al. 2008). Peptide immunotherapy using one of the GFAP peptide epitopes targeted by CD8+ T cells reduced the incidence of diabetes in this model (Tsui et al. 2008), reinforcing the concept that cytotoxic T cells are important in the destruction of the insulin-producing beta cells.
A second important question is whether similar autoreactive T cells are found in the pancreas, since most of the described autoreactive T cells have been identified using peripheral blood cells. This is a difficult issue, since it is difficult to obtain pancreata from recently-diagnosed T1D patients, but when these tissues have been available their analyses indicated that similar autoreactive T cell specificities can be identified. For example, for pre-proinsulin, DR4-restricted CD4+ T cell clones specific for the A-chain 1–15 epitope were the most dominant amongst the clones isolated from pancreatic lymph nodes (Kent et al. 2005); these were also detected in the peripheral blood of children with recently-diagnosed T1D (Marttila et al. 2008).
In the NOD mice, it has been shown that the frequency of CD8+ T cells specific for a peptide derived from IGRP in the peripheral blood could serve as a marker to identify mice that will develop disease. Interestingly, these IGRP-specific T cells have a high frequency amongst the islet-infiltrating T lymphocytes (Trudeau et al. 2003), where a substantial proportion of CD8+ T cells with a shared T cell receptor (TCR) α chain (Vα17-Jα42) recognize an IGRP-derived peptide. The pathogenic role of this prevalent T cell population is well established through the study of the 8.3 T cell clone, representative of this T cell population (Verdaguer et al. 1997).
From Antigen Identification to Epitope Discovery
As described above, several antigens have been identified as targets recognized by both T cells and autoantibodies in patients with T1D (Dudek and Purcell 2014; Han et al. 2013). New techniques such as the selective expression of beta-cell antigens using substraction microarrays have allowed for the identification of novel autoantigens recognized by CD4+ T cells despite the lack of a concurrent humoral immune responses (Neophytou et al. 1996; Roep and Peakman 2012). Interestingly, some of those autoantigens (i.e. chromogranin A: CHGA) are not only expressed by the pancreatic beta cells but also by cells of the neuroendocrine system. This led to the discovery of other targets expressed by non-beta cells in the islets, such as GFAP and S100beta.
However, the identification of the protein target represents only the first step in the characterization of the autoimmune T cell response. These cells are activated by peptide epitopes derived from proteins and bound to HLA molecules. Although it is possible to identify potential targets using algorithms predicting the affinity of those peptides for the corresponding HLA molecule, this strategy presents many caveats. Peptide epitopes with the highest binding scores for HLA molecules linked to the development of T1D are not always targeted by the T cell responses (Levisetti et al. 2008; Yang et al. 2014). Moreover, for peptide epitopes presented by HLA class II molecules flanking residues outside the binding groove are not predicted but have an important effect in the presentation/recognition of these epitopes (Rötzschke et al. 1999). In the case of HLA class I-restricted epitopes, there are algorithms that predict the COOH-terminus generated by either the proteasome or the immunoproteasome. Again, since the length of the presented peptide varies normally between 8 and 11 amino acids the exact epitope presented to T cells cannot be predicted. Finally, algorithms do not predict whether a peptide epitope is naturally processed and presented by an antigen-presenting cell in vivo.
These problems can be circumvented by the isolation and identification of the peptide epitopes present in the surface of antigen-presenting cells bound to HLA molecules. There are many examples that highlight the importance of this strategy in the discovery of peptide epitopes with clinical relevance in T1D. First, using a HLA-A*02:01/preproinsulin double-transfected K562 cell line as a surrogate beta cell, a new epitope derived from the pre-proinsulin (PPI) signal peptide was identified which was recognized by more than 50% of T1D patients (Skowera et al. 2008). In the case of MHC class II, S100b-derived peptide epitopes were discovered by incubating the lymphoblastoid cell line Priess (homozygous for the HLA class II DRB1*04:01 molecule) with the antigen. These epitopes were recognized more frequently by PBMCs from T1D patients compared to healthy donors (Gomez-Tourino et al. 2015), and this response was characterized by the secretion of IFN-γ. Interestingly, for some of this new S100b epitopes, significant responses were also detected in long-standing T1D patients suggesting that these S100b-specific cells could participate in the complications seen in these patients. More recently, by pulsing dendritic cells (DCs) with PPI, IA-2 and GAD65, the islet peptidome of high-risk MHC class II molecules DQ2, DQ8 and DQ2/8 has been published (van Lummel et al. 2016). These data indicate that the DQ2/8 molecule selectively presents peptide epitopes derived from PPI and IA-2 but not from GAD65. Moreover, one of the peptide epitopes from PPI lies in the signal peptide region of the molecule and has been previously described as a naturally processed epitope restricted by the HLA-A2.1 molecule (Skowera et al. 2008).
Despite the obvious generation of classical T cell peptide epitopes, recent work has suggested that post-translational modifications can be a new source of novel peptide autoantigens. This process has been extensively described in celiac disease, where T cell recognition of gluten antigens strongly depends in the posttranslational deamination of Gln residues (Qiao et al. 2011). Furthermore, the post-translational deimidation of Arg residues to citrulline has ben implicated in the generation of novel autoantigenic peptide epitopes in rheumatoid arthritis (Luban and Li 2010) and multiple sclerosis (Kidd et al. 2008). In T1D evidence of similar post-translational modifications generating novel antigenic peptide epitopes has been scarce although CD4 T cell clones capable of recognizing a post-translationally modified peptide epitope derived from the insulin A chain have been described. The recognition of this modified peptide epitope was dependent on the formation of a disulfide bond between adjacent Cys residues (Mannering et al. 2005).
More recent data suggests that the autoimmune responses in T1D can target hybrid peptides thus increasing the number of possible peptide epitopes (Delong et al. 2016). Hybrid peptides are formed by the fusion of insulin and CHGA peptides and are capable of stimulating CHGA-specific T cell clones, such as the well-known BDC-2.5. Moreover, those hybrid peptides are formed in vivo. CD4 T cells specific for these hybrid peptides were identified in both the spleen and pancreas of NOD mice, and more interestingly, similar autorreactive CD4 T cells were discovered in the pancreases of two T1D donors (Delong et al. 2016). This newly described posttranslational modification could represent an important source of new autoantigen peptide antigens.
What Is Different Between T1D Patients and Non-diabetic Subjects?
It has been debated for several years if there would be a difference between T1D patients and healthy subjects in terms of T cell responses against islet and non-islet antigens. Initial works suggested that in T1D patients, but not in healthy donors, autoreactive T cells escape thymic negative selection and are released in the peripheral blood; once in the periphery they get activated in draining lymph nodes by antigen-presenting cells displaying epitopes derived from islet antigens. This idea seems to be supported by the linkage of a genetic polymorphism in the PPI gene locus that determines the level of expression of this critical autoantigen in the thymus, which is related to the survival of PPI-specific T cells (Fan et al. 2009). It has been shown that depletion of the insulin-2 gene in medullary thymic epithelial cells was sufficient to break central tolerance to insulin. This generates an induce anti-insulin autoimmunity (Fan et al. 2009).
The idea that autoreactive T cells escape negative selection only in T1D patients stemmed from the fact that assays detecting T cell responses were limited. Furthermore, these assays, such as proliferation and thymidine incorporation assays, lacked sensitivity and were able to only detect responses in cases where the autoreactive T cells have been previously expanded in vivo. In addition, the generation of T cell lines by in vitro repetitive antigen stimulation of T cells was usually successful only in T1D patients, again probably due to the low precursor frequency in the peripheral blood of non-diabetic patients or the possibility that regulatory T (Treg) cells in T1D were defective (see below) (Brusko et al. 2005; Putnam et al. 2009). More recently, the use of nanoparticles coated with MHC molecules containing peptides derived from IGRP lead to the in vivo generation of CD4+ Treg-like cells (Tr1) and the reversal of established hyperglycemia in a mouse model (Clemente-Casares et al. 2016) showing that regulatory cells can be generated from antigen-experienced autoreactive lymphocytes. However, in the last few years, new analytical techniques, such as ELISPOT or the use of HLA tetrameric molecules, have changed this paradigm. Many groups have shown that it is possible to detect autoreactive T cells in the peripheral blood of healthy donors (Yu et al. 2015), suggesting that regulatory circuits might be defective in T1D patients and thus allow the activation and expansion of these cells. Recent studies show the difference in T1D patients versus healthy donors is the frequency and, more importantly, the phenotype of such autoreactive T lymphocytes, especially in the case of CD4+ T cells. For example, islet-specific CD4+ T cells show a pro-inflammatory phenotype in T1D patients, characterized by a skewed Th1 phenotype, when compared to the more regulatory phenotype in healthy donors (Arif et al. 2004). New incoming technologies, such as multiplex and single-cell PCR and TCR clonotyping, will push this field further by analyzing in depth the phenotype of each of these autoreactive T cells and their specificity (Eugster et al. 2013; Gomez-Tourino et al. 2016).
Regulation of the Autoinmune Response in T1D: Targeting Effector T Cells
The basic concept regarding the origin of the autoimmune response is that several defects in the innate and adaptative branch of the immune system lead to an imbalance in the regulation of the response allowing the activation and expansion of pathogenic effector T and B lymphocytes. Several interventions have been developed to restore homeostasis in these patients; the use of cyclosporine, prednisone, anti-CD3 antibodies, anti-thymocyte globulin or autologous hematopoietic stem cell transplantion are examples of those therapies (Peakman and von Herrath 2010). Critically, one of the main problems of these strategies is their non-specificity. It is now evident that Treg cells are in the control of any autoimmune response. The characterization of Treg cells and their generation in vitro and in vivo has opened a whole new field in the treatment of autoimmune diseases.
The first subset of Tregs to be definied are CD4+CD25+CD127lo/−FoxP3+ T lymphocytes (Putnam et al. 2009); work in the NOD mice regarding this T cell population has been controversial. Initial studies have suggested a primary defect in the number of these cells (Alard et al. 2006) but these results not reproducible by other groups (Mellanby et al. 2007). These discrepancies probably arise from differences between studies regarding the identity of Treg cells. However, recent work suggest that differences between diabetes-prone NOD mice and other mice strains may reside in Treg function. Mechanisms, including age-related decline of transforming growth factor (TGF)-β production (Gregg et al. 2004), interleukin (IL)-2 insufficiency (Yamanouchi et al. 2007), resistance of effector T cells to regulation (D’Alise et al. 2008), or an unusually reduced TCR diversity (Ferreira et al. 2014), are often cited to explain these differences in animal models.
In humans, an initial report indicated a reduced frequency of CD4+CD25+ cells in both newly diagnosed and long-standing T1D patients (Kukreja et al. 2002). However, subsequent studies did not find this reduction (Brusko et al. 2005; Kriegel et al. 2004; Lindley et al. 2005; Putnam et al. 2005). Regarding the suppressive activity of the Treg population results are quite variable: functional abnormalities were reported in two studies (Brusko et al. 2005; Lindley et al. 2005). Despite these conflicting results, recent clinical trials indicates that ex vivo-expanded autologous Treg from T1D patients are functional both in vivo once administered into patients and in vitro (Bluestone et al. 2015; Marek-Trzonkowska et al. 2012, 2014).
A new subgroup of CD4+ population of Treg cells have been identified, the so-called type 1 regulatory T (Tr1) cells (Groux et al. 1997). Tr1 cells are characterized by the secretion of high levels of IL-10, TGF-β, a lack of expression of FoxP3, and their capacity to suppress immune responses. These cells are antigen-specific and can be generated both in vitro and in vivo using several strategies (Zeng et al. 2015). Several studies have shown their importance in the regulation of the autoimmune response in T1D. Treatment of diabetic mice with IL-10 and rapamycin after islet transplantation induced stable long-term islet tolerance (Battaglia et al. 2006). IL-10-secreting Tr1 cells suppressed T cell proliferation and transferred antigen-specific tolerance to mice receiving new transplants (Battaglia et al. 2006). Tr1 cells were also detected in T1D patients after pancreatic islet transplantation in insulin-independent recipients (Huurman et al. 2009). More recently it has been shown that it is possible to generate to antigen-specific Tr1 cells in vivo. Systemic delivery of nanoparticles coated with class II HLA molecules containing disease-relevant peptides triggers the generation of Tr1 cells in NOD mice and restored normoglycaemia in a high percentage of diabetic mice (Clemente-Casares et al. 2016). These data open up the possibility of antigen-specific immunotherapy in human patients capable of regulating autoimmune responses.
Cytotoxic CD8 T Lympochytes: Are They Regulatory?
Usually, cytotoxic T cells in T1D are noted for their potential role in the destruction of the beta cells in the pancreas. Although data supporting this notion has been scarce, recent work show that this T cell subset is present at a high frequency in insulitic lesions and in the peripheral blood of T1D patients. The presence of CD8+ T cells and MHC class I hyperexpression in frozen pancreas tissue samples has been demonstrated in both newly-diagnosed and long-standing T1D patients. The use of in situ tetramer staining shows that some of these cells are specific for islet antigens (Coppieters et al. 2012). A recent study using flow cytometry and tetramer staining shows that the frequency of beta-cell-specific CD8 T cells is similar between T1D patients and healthy donors; however, in patients, molecular analysis shows highly skewed oligoclonal TCR repertoires, a sign of chronic autoantigen exposure (Skowera et al. 2015). Interestingly, indirect evidence on the possible role of CD8+ T cells in the destruction of insulin-producing beta-cells in vivo comes from the results of a recent clinical trial: the C-peptide levels in T1D patients injected with a proinsulin-encoding plasmid improved when compared to placebo. Moreover, proinsulin-specific CD8+ T cells, but not T cells specific for other islet autoantigens, were reduced in peripheral blood of these patients. However, no significant changes were detected in cytokine production by CD4+ T cells, indicating a critical role for cytotoxic T cells in T1D (Roep et al. 2013). This beta-cell specific cytolysis by CD8+ T cells is supported by in vitro studies of CD8+ T cell clones specific for islet autoantigens (Skowera et al. 2008). Since the identification of a novel peptide epitope derived from the signal peptide of PPI, PPI-specific CD8+ T cells were subsequently shown to be cytotoxic against both the corresponding peptide- and whole PPI-pulsed cells. Furthermore, they were also cytotoxic in vitro against beta cells of isolated islets obtained from organ donors. This cytotoxicity was specific for the beta cells, since (1) glucagon-producing alpha cells in the same islets remained intact and (2) only islets expressing the MHC class I molecule A*02:01, the molecule presenting the peptide to these CD8+ T cell clones, were lysed (Skowera et al. 2008).
Although much of the data regarding the specific role of CD8+ T cells in the destruction of the beta cells relates to the role of proinsulin-specific cytotoxic T cells, it has also been shown that other autoantigen-specific CD8+ T cells are capable of destroying beta cells. The transfer of a MHC class I A*02:01-restricted IGRP-specific CD8+ T cell clone derived from a T1D patient into the HLA-A2 NOD-SCID mouse, triggered destructive insulitis and diabetes in vivo (Unger et al. 2012).
It is also known that the CD8+ T cell subset as a whole is heterogeneous and includes more cellular phenotypes, apart from the prototypical cytotoxic role (Mittrücker et al. 2014). Interestingly, in the NOD mice, CD8+ T cells specific for islet antigens may have a regulatory role that could delay/inhibit the development of T1D. The first report suggesting a regulatory role for this T cell subset was published in the mid-70s (Cantor et al. 1976). However, their role in immune regulation was soon superseded by the discovery of CD25-expressing CD4+ T cells as the major T cell subset involved in the regulation of the immune response (Sakaguchi et al. 1995).
The regulatory role played by CD8+ T cells was first proposed by data from mice where this cell subset (restricted by the Qa-1 molecule—the homologue of the human HLA-E molecule) limited or regulated many types of immune responses in vivo, including autoimmune diseases, such as experimental allergic encephalomyelitis (Jiang et al. 1998). In T1D, the presence of these HLA-E-restricted CD8+ Treg cells has also been demonstrated. It has been reported that regulatory CD8+ T cells specific for an epitope derived from the signal sequence of the heat shock protein (HSP)60 can be detected in the peripheral blood (Jiang et al. 2010). More importantly, those cells do not seem to be effective in patients with T1D and could be generated in vitro by immature DCs pulsed with this HSP peptide.
It has also been reported that CD8+ T cells specific for peptide epitopes derived from autoantigens expressed by islet cells can be differentiated into regulatory T cells (Tsai et al. 2010). Immunization of NOD mice transgenic for a TCR derived from a diabetogenic CD8+ T cell, specific for an IGRP peptide, led to the differentiation of CD8+ T cells with an autoregulatory phenotype (Tsai et al. 2010). These memory-like autoregulatory T cells not only prevented the development of T1D in prediabetic animals, but also restored normoglycemia in diabetic animals. The mechanism proposed by which these cells operate seems to be by deletion of antigen-presenting cells bearing the specific peptide in the cell surface in a perforin-dependent mechanism. Moreover, in a polyclonal T cell animal model expressing a human class I HLA molecule, two different nanoparticle systems bearing peptides derived from either IGRP or PPI were capable of generating memory-like autoregulatory CD8 T cells and restore normoglycemia in a high percentage of treated animals (Tsai et al. 2010).
Conclusions
In this review we describe the role of T cell responses in T1D. Currently, many autoantigens recognized by these cells have been identified, including antigens expressed by non-islet cells, such as the neurotropic factor S100b or GFAP; specific peptide epitopes derived from those autoantigens have been discovered and T-cell responses against these have been quantified in both T1D patients and healthy donors. These results indicate that a qualitative difference in the T cell response between both groups of subjects and probably in the regulation of the T cell response by Treg cells.
However, recently, the role of CD8+ T cells in the development of the disease has been intensively studied raising two critical questions. First, the generation of islet-specific cytotoxic T cells appears to be an important effector mechanism leading to the destruction of the beta cells; and second, the same cells could play an important role in preventing the development of clinical symptoms by their differentiation into memory-like autoregulatory T cells, a differentiation route that can be enforced using nanoparticle vaccines. This dual role played by CD8+ T cells could be used to prevent the development of the disease in T1D patients in the future.
References
Alard P, Manirarora JN, Parnell SA et al (2006) Deficiency in NOD antigen-presenting cell function may be responsible for suboptimal CD4+ CD25+ T-cell-mediated regulation and type 1 diabetes development in NOD mice. Diabetes 55:2098–2105
Anderson MS, Bluestone JA (2005) The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 23:447–485
Arif S, Tree TI, Astill TP et al (2004) Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest 113:451–463
Arif S, Leete P, Nguyen V et al (2014) Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes 63:3835–3845
Atkinson MA (2012) The pathogenesis and natural history of type 1 diabetes. Cold Spring Harb Perspect Med 2 pii:a007641
Babad J, Geliebter A, DiLorenzo TP (2010) T-cell autoantigens in the non-obese diabetic mouse model of autoimmune diabetes. Immunology 131:459–465
Banwell B, Bar-Or A, Cheung R et al (2008) Abnormal T-cell reactivities in childhood inflammatory demyelinating disease and type 1 diabetes. Ann Neurol 63:98–111
Battaglia M, Stabilini A, Draghici E et al (2006) Rapamycin and interleukin-10 treatment induces T regulatory type 1 cells that mediate antigen-specific transplantation tolerance. Diabetes 55:40–49
Bluestone JA, Buckner JH, Fitch M et al (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7:315ra189
Brusko TM, Wasserfall CH, Clare-Salzler MJ et al (2005) Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes 54:1407–1414
Cantor H, Shen FW, Boyse EA (1976) Separation of helper T cells from suppressor T cells expressing different Ly components. II. Activation by antigen: after immunization, antigen-specific suppressor and helper activities are mediated by distinct T-cell subclasses. J Exp Med 143:1391–1340
Clemente-Casares X, Blanco J, Ambalavanan P et al (2016) Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 530:434–440
Concannon P, Erlich HA, Julier C et al (2005) Type 1 diabetes: evidence for susceptibility loci from four genome-wide linkage scans in 1,435 multiplex families. Diabetes 54:2995–3001
Concannon P, Chen WM, Julier C et al (2009) Genome-wide scan for linkage to type 1 diabetes in 2,496 multiplex families from the Type 1 diabetes genetics consortium. Diabetes 58:1018–1022
Coppieters KT, Dotta F, Amirian N et al (2012) Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med 209:51–60
D’Alise AM, Auyeung V, Feuerer M et al (2008) The defect in T-cell regulation in NOD mice is an effect on the T-cell efectors. Proc Natl Acad Sci USA 105:19857–19862
Dabelea D, Mayer-Davis EJ, Saydah S et al (2014) Prevalence of type 1 and type 2 diabetes among children and adolescents from 2001 to 2009. JAMA 311:1778–1786
Delong T, Wiles TA, Baker RL et al (2016) Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 351:711–714
Di Lorenzo TP, Peakman M, Roep BO (2007) Translational mini-review series on type 1 diabetes: systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol 148:1–16
Dudek NL, Purcell AW (2014) The beta cell immunopeptidome. Vitam Horm 95:115–144
Erlich H, Valdes AM, Noble J et al (2008) HLA DR-DQ haplotypes and genotypes and type 1 diabetes risk: analysis of the type 1 diabetes genetics consortium families. Diabetes 57:1084–1092
Eugster A, Lindner A, Heninger AK et al (2013) Measuring T cell receptor and T cell gene expression diversity in antigen-responsive human CD4 + T cells. J Immunol Methods 400–401:13–22
EURODIAB ACE Study Group (2000) Variation and trends in incidence of childhood diabetes in Europe. Lancet 355:873–876
Fan Y, Rudert WA, Grupillo M et al (2009) Thymus-specific deletion of insulin induces autoimmune diabetes. EMBO J 28:2812–2824
Ferreira C, Pallmer D, Blake K et al (2014) Reduced regulatory T cell diversity in NOD mice is linked to early events in the thymus. J Immunol 192:4145–4152
Foulis AK, McGill M, Farquharson MA (1991) Insulitis in type 1 (insulin-dependent) diabetes mellitus in man–macrophages, lymphocytes, and interferon-gamma containing cells. J Pathol 165:97–103
Gomez-Tourino I, Camina-Darriba F, Otero-Romero I et al (2010) Autoantibodies to glial fibrillary acid protein and S100beta in diabetic patients. Diabet Med 27:246–248
Gomez-Tourino I, Simon-Vazquez R, Alonso-Lorenzo J et al (2015) Characterization of the autoimmune response against the nerve tissue S100beta in patients with type 1 diabetes. Clin Exp Immunol 180:207–217
Gomez-Tourino I, Arif S, Eichmann M et al (2016) T cells in type 1 diabetes: instructors, regulators and effectors: a comprehensive review. J Autoimmun 66:7–16
Gregg RK, Jain R, Schoenleber SJ et al (2004) A sudden decline in active membrane-bound TGF-beta impairs both T regulatory cell function and protection against autoimmune diabetes. J Immunol 173:7308–7316
Groux H, O’Garra A, Bigler M et al (1997) A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature 389:737–742
Han S, Donelan W, Wang H et al (2013) Novel autoantigens in type 1 diabetes. Am J Transl Res 5:379–392
Hanafusa T, Imagawa A (2008) Insulitis in human type 1 diabetes. Ann N Y Acad Sci 1150:297–299
Huurman VA, Velthius JH, Hilbrands R et al (2009) Allograft-specific cytokine profiles associate with clinical outcome after islet transplantation. Am J Transplant 9:382–388
International Diabetes Federation (2015) International Diabetes Federation Atlas. http://www.diabetesatlas.org. Accessed 07 Apr 2016
Itoh N, Hanafusa T, Miyazaki A et al (1993) Mononuclear cell infiltration and its relation to the expression of major histocompatibility complex antigens and adhesion molecules in pancreas biopsy specimens from newly diagnosed insulin-dependent diabetes mellitus patients. J Clin Invest 92:2313–2322
Jiang H, Kashleva H, Xu LX et al (1998) T cell vaccination induces T cell receptor Vbeta-specific Qa-1-restricted regulatory CD8(+) T cells. Proc Natl Acad Sci USA 95:4533–4537
Jiang H, Canfield SM, Gallagher MP et al (2010) HLA-E-restricted regulatory CD8(+) T cells are involved in development and control of human autoimmune type 1 diabetes. J Clin Invest 120:3641–3650
Kent SC, Chen Y, Bregoli L et al (2005) Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature 435:224–228
Kidd BA, Ho PP, Sharpe O et al (2008) Epitope spreading to citrullinated antigens in mouse models of autoimmune arthritis and demyelination. Arthritis Res Ther 10:R119
Koo BK, Chae S, Kim KM et al (2014) Identification of novel autoantibodies in type 1 diabetic patients using a high-density protein microarray. Diabetes 63:3022–3032
Kriegel MA, Lohmann T, Gabler C et al (2004) Defective suppressor function of human CD4+ CD25+ regulatory T cells in autoimmune polyglandular syndrome type II. J Exp Med 199:1285–1291
Kukreja A, Cost G, Marker J et al (2002) Multiple immuno-regulatory defects in type-1 diabetes. J Clin Invest 109:131–140
Leete P, Willcox A, Krogvold L et al (2016) Differential insulitic profiles determine the extent of beta cell destruction and the age at onset of type 1 diabetes. Diabetes 65:1362–1369
Levisetti MG, Lewis DM, Suri A et al (2008) Weak proinsulin peptide-major histocompatibility complexes are targeted in autoimmune diabetes in mice. Diabetes 57:1852–1860
Lieberman SM, Evans AM, Han B et al (2003) Identification of the beta cell antigen targeted by a prevalent population of pathogenic CD8+ T cells in autoimmune diabetes. Proc Natl Acad Sci USA 100:8384–8388
Lindley S, Dayan CM, Bishop A et al (2005) Defective suppressor function in CD4+ CD25+ T-cells from patients with type 1 diabetes. Diabetes 54:92–99
Luban S, Li ZG (2010) Citrullinated peptide and its relevance to rheumatoid arthritis: an update. Int J Rheum Dis 13:284–287
Mannering SI, Harrison LC, Williamson NA et al (2005) The insulin A-chain epitope recognized by human T cells is posttranslationally modified. J Exp Med 202:1191–1197
Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A et al (2012) Administration of CD4+ CD25highCD127-regulatory T cells preserves b-cell function in type 1 diabetes in children. Diabetes Care 35:1817–1820
Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A et al (2014) Therapy of type 1 diabetes with CD4+ CD25highCD127-regulatory T cells prolongs survival of pancreatic islets—results of 1 year follow-up. Clin Immunol 153:23–30
Marron MP, Graser RT, Chapman HD et al (2002) Functional evidence for the mediation of diabetogenic T cell responses by HLA-A2.1 MHC class I molecules through transgenic expression in NOD mice. Proc Natl Acad Sci USA 99:13753–13758
Marttila J, Huttunen S, Vaarala O et al (2008) T-cell reactivity to insulin peptide A1-12 in children with recently diagnosed type 1 diabetes or multiple beta-cell autoantibodies. J Autoimmun 31:142–148
Mellanby RJ, Thomas D, Phillips JM et al (2007) Diabetes in non-obese diabetic mice is not associated with quantitative changes in CD4+ CD25+ Foxp3+ regulatory T cells. Immunology 121:15–28
Mittrücker HW, Visekruna A, Huber M (2014) Heterogeneity in the differentiation and function of CD8+ T cells. Arch Immunol Ther Exp 62:449–458
Neophytou PI, Roep BO, Arden SD et al (1996) T-cell epitope analysis using subtracted expression libraries (TEASEL): application to a 38-kDA autoantigen recognized by T cells from an insulin-dependent diabetic patient. Proc Natl Acad Sci USA 93:2014–2018
Ouyang Q, Standifer NE, Qin H et al (2006) Recognition of HLA class I-restricted beta-cell epitopes in type 1 diabetes. Diabetes 55:3068–3074
Peakman M, von Herrath M (2010) Antigen-specific immunotherapy for type 1 diabetes: maximizing the potential. Diabetes 59:2087–2093
Putnam AL, Vendrame F, Dotta F et al (2005) CD4 + CD25high regulatory T cells in human autoimmune diabetes. J Autoimmun 24:55–62
Putnam AL, Brusko TM, Lee MR et al (2009) Expansion of human regulatory T-cells from patients with type 1 diabetes. Diabetes 58:652–662
Qiao SW, Ráki M, Gunnarsen KS et al (2011) Posttranslational modification of gluten shapes TCR usage in celiac disease. J Immunol 187:3064–3071
Roep BO, Peakman M (2012) Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med 2:a007781
Roep BO, Solvason N, Gottlieb PA et al (2013) Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8(+) T cells in type 1 diabetes. Sci Transl Med 5:191ra82
Rötzschke O, Falk K, Mack J et al (1999) Conformational variants of class II MHC/peptide complexes induced by N- and C-terminal extensions of minimal peptide epitopes. Proc Natl Acad Sci USA 96:7445–7450
Sakaguchi S, Sakaguchi N, Asano M et al (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155:1151–1164
Serre L, Fazilleau N, Guerder S (2015) Central tolerance spares the private high-avidity CD4(+) T-cell repertoire specific for an islet antigen in NOD mice. Eur J Immunol 45:1946–1956
Skowera A, Ellis RJ, Varela-Calvino R et al (2008) CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose-regulated preproinsulin epitope. J Clin Invest 118:3390–3402
Skowera A, Ladell K, McLaren JE et al (2015) Beta-cell-specific CD8 T cell phenotype in type 1 diabetes reflects chronic autoantigen exposure. Diabetes 64:916–925
Standifer NE, Ouyang Q, Panagiotopoulos C et al (2006) Identification of novel HLA-A*0201-restricted epitopes in recent-onset type 1 diabetic subjects and antibody-positive relatives. Diabetes 55:3061–3067
Trudeau JD, Kelly-Smith C, Verchere CB et al (2003) Prediction of spontaneous autoimmune diabetes in NOD mice by quantification of autoreactive T cells in peripheral blood. J Clin Invest 111:217–223
Tsai S, Shameli A, Yamanouchi J et al (2010) Reversal of autoimmunity by boosting memory-like autoregulatory T cells. Immunity 32:568–580
Tsui H, Chan Y, Tang L et al (2008) Targeting of pancreatic glia in type 1 diabetes. Diabetes 57:918–928
Unger WW, Pearson T, Abreu JR et al (2012) Islet-specific CTL cloned from a type 1 diabetes patient cause beta-cell destruction after engraftment into HLA-A2 transgenic NOD/scid/IL2RG null mice. PLoS One 7:e49213
van Lummel M, van Veelen PA, de Ru AH et al (2016) Discovery of a selective islet peptidome presented by the highest-risk HLA-DQ8trans molecule. Diabetes 65:732–741
Verdaguer J, Schmidt D, Amrani A et al (1997) Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J Exp Med 186:1663–1676
Verge CF, Gianani R, Kawasaki E et al (1996) Prediction of type I diabetes in first-degree relatives using a combination of insulin, GAD, and ICA512bdc/IA-2 autoantibodies. Diabetes 45:926–933
Wenzlau JM, Hutton JC (2013) Novel diabetes autoantibodies and prediction of type 1 diabetes. Curr Diab Rep 13:608–615
Wenzlau JM, Juhl K, Yu L et al (2007) The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci USA 104:17040–17045
Wenzlau JM, Moua O, Sarkar SA et al (2008) SlC30A8 is a major target of humoral autoimmunity in type 1 diabetes and a predictive marker in prediabetes. Ann N Y Acad Sci 1150:256–259
Willcox A, Richardson SJ, Bone AJ et al (2009) Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol 155:173–181
Winer S, Tsui H, Lau A et al (2003) Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 9:198–205
Winer DA, Winer S, Shen L et al (2011) B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat Med 17:610–617
Yamanouchi J, Rainbow D, Serra P et al (2007) Interleukin-2 gene variation impairs regulatory T cell function and causes autoimmunity. Nat Genet 39:329–337
Yang J, Chow IT, Sosinowski T et al (2014) Autoreactive T cells specific for insulin B:11–23 recognize a low-affinity peptide register in human subjects with autoimmune diabetes. Proc Natl Acad Sci USA 111:14840–14845
Yu W, Jiang N, Ebert PJ et al (2015) Clonal Deletion prunes but does not eliminate self-specific alphabeta CD8(+) T lymphocytes. Immunity 42:929–941
Zeng H, Zhang R, Jin B et al (2015) Type 1 regulatory T cells: a new mechanism of peripheral immune tolerance. Cell Mol Immunol 12:566–571
Acknowledgements
This work was supported by a Grant of RETOS Program by the Spanish Ministerio de Economía y Competitividad (MINECO), co-funded with FEDER funds (Grant 2014-PN215). We would like to acknowledge Sefina Arif for critically reviewing the manuscript.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Varela-Calvino, R., Calviño-Sampedro, C., Gómez-Touriño, I. et al. Apportioning Blame: Autoreactive CD4+ and CD8+ T Cells in Type 1 Diabetes. Arch. Immunol. Ther. Exp. 65, 275–284 (2017). https://doi.org/10.1007/s00005-016-0452-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00005-016-0452-4