
Abstract
Progesterone (PRG) and testosterone (TST) were impregnated on mesoporous silica (ExP) particles via supercritical carbon dioxide (scCO2) processing at various pressures (10–18 MPa), temperatures (308.2–328.2 K), and time (30–360 min). The impact of a co-solvent on the impregnation was also studied at the best determined pressure and temperature. The properties of the drug embedded in silica particles were analysed via gas chromatography (GC), attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy, X-ray diffraction (XRD), differential scanning calorimetry (DSC), and nitrogen adsorption. An impregnation of 1 to 82 mg/g for PRG and 0.1 to 16 mg/g for TST was obtained depending on the processing parameters. There was a significant effect of pressure, time, and co-solvent on the impregnation efficiency. Generally, an increase in time and pressure plus the use of co-solvent led to an improvement in drug adsorption. Conversely, a rise in temperature resulted in lower impregnation of both TST and PRG on ExP. There was a substantial increase in the dissolution rate (> 90% drug release within the first 2 min) of both TST and PRG impregnated in silica particles when compared to the unprocessed drugs. This dissolution enhancement was attributed to the amorphisation of both drugs due to their adsorption on mesoporous silica.
Graphical Abstract

Similar content being viewed by others

Introduction
The poor aqueous solubility and dissolution rate of certain active therapeutic ingredients are still part of the major challenges in pharmaceutical development [1]. When delivered orally, hydrophobic drugs tend to show a dissolution-limited absorption and low bioavailability [2]. Therefore, it is crucial that strategies are designed and developed to help overcome the solubility issues of these problematic molecules. Several preparation techniques for the improvement of drug solubility have been evaluated in research including micro-/nano-emulsions [3, 4], micelles [5], complexation [6], particle size reduction [7], prodrug or salt formation [8, 9], solid lipid nanoparticles [10], and immobilisation onto porous carriers [11]. Generally, it has been recognised that increasing the specific surface area of a poorly water-soluble drug in contact with the dissolution medium could result in a better drug solubility profile [12] [12]. The impregnation of a therapeutic molecule onto mesoporous particles can improve the dissolution rate by increasing the surface area as well as due to the stable (pore network restricts drug re-crystallisation) drug amorphisation [13, 14].
The mesoporous silica particles for drug delivery are highly attractive due to their desirable features including biocompatibility, non-toxicity, high surface area, tuneable pore sizes, and stable and uniform pore structures [15, 16]. Also, the surface of the solid carrier comprises free hydroxyl groups which are easily accessible for specific interactions (e.g. hydrogen bonding) with adsorbing molecules [17]. Mesoporous silica nanoparticles have been greatly applied as drug delivery vehicles in developing the treatment of several clinical disorders including diabetes [18, 19], cancer [20,21,22], hypertension [23], depression [24], bacterial infections [25], and osteoporosis [26, 27]. The use of delivery systems based on mesoporous silica allows for modifications to enable a controlled release of payload in response to internal/external stimuli such as pH, light, temperature, and glucose. Several clinical studies in humans have shown evidence of the safety and efficacy of silica nanoparticles [28]. In one trial with 16 healthy adult participants, the oral delivery of ibuprofen via silica-lipid hybrid systems led to a 1.95 times improvement in bioavailability [29]. Another study by Bukara et al. indicated the potential of ordered mesoporous silica to enhance the oral bioavailability of poorly water-soluble fenofibrate in 12 healthy volunteers, compared to a commercially marketed micronised fenofibrate formulation [30]. Similarly, the trials on the use of ultrafine silica particles (7 nm) such as Cornell-dots (C-dots) for potential cancer imaging in metastatic melanoma or malignant brain tumours are currently ongoing [31]. Although there are still obstacles to overcome concerning the clinical translation of silica nanoparticles (e.g. long-term toxicology profile), their aforementioned advantageous structural and physicochemical properties still make them suitable contenders as delivery vehicles. Also, their relatively higher stability in harsh conditions including the acidic gastric environment compared to other conventional systems like liposomes makes them highly attractive for the oral delivery of sensitive biotherapeutics [28, 32, 33]. It has been shown that therapeutics of different sizes can be immobilised within the mesopores using an appropriate impregnation method and desorbed through a diffusion-controlled mechanism [34, 35]. Various impregnation techniques for hydrophobic drugs on silica have been explored in literature which typically involve physical mixing [36], and solvent-based methods where loading is done by either a dropwise addition of concentrated drug solution [37,38,39] or suspension of silica into the drug-solvent solution [40, 41]. The drug loading procedures into the mesopores of silica have been recently reviewed by Trzeciak et al. and Seljak et al. [42, 43]. However, these established methods usually present challenges that could restrict their use. For example, chemical/thermal degradation or residual solvent toxicity remains a concern with the preparation of formulations via solvent-based processes [44]. Therefore, it is important to explore alternative processing techniques, such as supercritical fluid (SCF) processing, which has the potential to avoid most of these drawbacks [45].
SCF can be described as a substance above its critical pressure and temperature, where it possesses density and diffusivity properties like liquid and gas, respectively [46]. Moreover, these physicochemical properties of SCF can be tuned by varying the operating pressure and temperature [46, 47]. Supercritical carbon dioxide (scCO2) is one of the most commonly used SCF because of its low critical temperature (304.3 K) and pressure (7.38 MPa). CO2 is also inert, non-flammable, non-toxic, and readily available [47]. The tuneable properties of scCO2 make it very adaptable in pharmaceutical processing as it can be applied as an extraction agent, anti-solvent, solvent, and/or plasticiser for various drugs and polymers [48,49,50,51]. The scCO2 has found its role as a substitute for organic solvents in the impregnation of drugs onto porous supports. Under scCO2 conditions, the adsorption process is typically a single-step method where the initial stage involves pressurisation of CO2 to allow for considerable drug solubilisation and easy dispersion of the solute throughout the adsorbing mesopore network. Following depressurisation, the scCO2 is released, thus causing the solubilised drug to precipitate and remain within the porous matrix [52, 53]. A co-solvent is important with processing materials that are sparingly soluble in scCO2. For these instances, after the impregnation procedure, the scCO2 flow is sustained for an adequate time to allow for the complete removal of the solvent [54]. The impregnation of different drugs on porous silica via SCF processing has already been reported in the literature, and a few of those examples are listed in Table I.
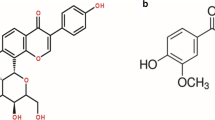
In this work, scCO2 was employed for the impregnation of progesterone (PRG) and testosterone (TST) onto mesoporous silica particles. TST (Fig. 1a) and PRG (Fig. 1b) are known as endogenous steroids, and they play a significant role in sexuality and fertility.

Chemical structures of testosterone (a) and progesterone (b)
The poor aqueous solubility of these sex hormones (approximately 10 mg/mL) limits their absorption and bioavailability after oral intake [68, 69]. Both TST and PRG are classified as class II according to the Biopharmaceutics Classification System (BCS) and class IIb as per the Developability Classification System (DCS) [70]. Until now, there have been several studies to improve the dissolution of PRG and TST, including the formation of cocrystals [71], cyclodextrin complexation [72,73,74,75,76], polymeric nanoparticles [77, 78], micro-/mesoporous materials [79,80,81], and micronisation via rapid expansion of supercritical solution [82]. The supercritical impregnation process is a simple technique with several advantages over conventional methods, including (i) homogenous distribution of active substance within the solid matrix, (ii) environmentally friendly technique for the reduction of waste and the use of toxic organic solvents, (iii) shorter processing times as there is no requirement for an additional drying step, and (iv) any excess drug active can be recycled by avoiding cross-contamination [83]. The scCO2 processing parameters such as pressure, temperature, time, and the use of a co-solvent have a strong impact on the solubility and impregnation efficiency of hydrophobic drugs. The solubility behaviour of PRG and TST in scCO2 has already been detailed in the literature [82, 84, 85]. However, TST and PRG impregnation on mesoporous silica via scCO2 processing has not been reported previously. Thus, the current study aims to investigate the influence of processing parameters on the impregnation efficiency of both steroids onto silica with the view of exploring the scCO2 technique as a viable option in developing TST and PRG formulations with improved aqueous solubility. Thereafter, the physical properties of selected drug-loaded particles were characterised using ATR-FTIR, XRD, SEM, DSC, and nitrogen adsorption. The dissolution rate of PRG and TST from the loaded silica particles was also investigated in this study.
Materials and Methods
Materials
Progesterone (> 99% pure; Pharmacia & Upjohn, USA), testosterone (> 99% pure; Pharmacia & Upjohn, USA), Mesoporous Exmere Plus silica (average particle size: 9 mm, specific surface area: 281 m2/g, pore diameter: 19.6 nm; Exmere Ltd., UK), Grace-9396 (9396) silica particles (average particle size: 200 mm, pore diameter: 450 nm; W.R. Grace & Co, USA), tetrahydrofuran (> 99.5% pure; ACS grade, Thermo Fisher, UK), and liquid CO2 (99.9% pure; BOC Ltd., UK). All other chemicals used in this work were of analytical grade and used without further purification.
Methods
scCO2 Impregnation of PRG and TST onto Silica
The scCO2 processing was carried out in the static mode to obtain PRG/TST-loaded silica using an apparatus supplied by Thar Process Inc., Pittsburgh, PA, USA, as described in detail elsewhere [86]. For drug loading experiments in scCO2, known quantities of the TST or PRG and a fixed amount of the mesoporous silica (700 mg) were sealed separately in a porous filter paper (85 mm pore size; Fisher Scientific, UK) to minimise the direct contact between both materials. Then, the bags were placed into a beaker and transferred inside the high-pressure vessel pre-heated to 208.2 or 328.2 K (± 2 K). For experiments involving the use of co-solvent, 10 mL of tetrahydrofuran (THF) was introduced into the bottom of the vessel before placing the beaker in the vessel. Liquid CO2 was pumped from a cylinder via a cooling unit into the vessel to the desired pressure (10 to 18 MPa). The vessel content was then allowed to equilibrate for a specified time between 30 and 360 min at a constant pressure achieved via an automatic back-pressure regulator. Afterwards, the vessel was slowly depressurized at a rate of 1.2 MPa/min and the drug-adsorbed particles were recovered. The amount of PRG or TST loaded onto silica (mg/g) was measured by solubilising a known amount of the formulation into methanol (MeOH), followed by concentration analysis using ultraviolet–visible (UV–Vis) spectroscopy (Cary 3500 UV–Vis spectrometer, Agilent Technologies, UK) at 240 nm.
Characterisation
The residual solvent in the samples prepared in the presence of THF was determined by gas chromatography (GC). An Agilent 6850 GC system (Agilent Technologies, Waldbronn, Germany) was used to perform the analysis with a liquid autosampler. Samples were introduced in a split/splitless injection port to a HP-1 (30 m length × 0.32 mm i.d., 0.25 μm film thickness) column and detection was performed using a flame ionisation detector (FID). The column oven was programmed with an initial column oven temperature of 303.2 K and held at the same temperature for the total run time of 6 min. The injector and detector temperatures were kept at 473.2 K and 523.2 K, respectively. Helium was used as a carrier gas with a head pressure of 0.054 MPa resulting in an initial column flow of 3.2 mL/min and an average velocity of 50 cm/s. Helium was also used as a makeup gas for the FID detector. The makeup gas flow rate was 5 mL/min, whilst for hydrogen and nitrogen the flow was 5 mL/min and 10 mL/min, respectively. Samples were injected by the instrument’s autosampler with an injection volume of 1.0 μL, and deionised water was used to rinse the syringe between injections. The residual solvent in the sample was determined with the help of a previously prepared calibration curve of THF in ethanol at 10, 20, 30, 40, and 50 mg/mL concentrations. For the residual solvent analysis, 100 mg of drug-loaded silica was dispersed in 10 mL of ethanol in a closed vial and sonicated for 30 min prior to the injection in GC.
DSC analysis of PRG, TST, and drug-loaded ExP particles was performed using a DSC823e calorimeter (Mettler Toledo, LLC, Leicester, UK). For each run, 3 to 4 mg of sample were weighed and hermetically sealed in the aluminium pan. The sealed pans were then heated at a rate of 10 K/min after placing them in the DSC. The DSC data was collected under a constant flow of nitrogen (40 mL/min) over the temperature range of 295 to 475 K.
The attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra of the drug and free/loaded silica were obtained using a Spectrum Two FTIR spectrophotometer (Perkin Elmer, UK). Approximately 1 to 2 mg of the sample were uniformly spread on the surface of a single reflection horizontal ATR accessory with a zinc selenide (ZnSe) crystal. The spectra were collected in the transmission mode over the range of 4000–400 cm−1. Each spectrum was comprised of 16 scans with a resolution of 8 cm−1.
The X-ray diffraction (XRD) analysis on PRG, TST, and drug-adsorbed ExP particles was conducted using a Bruker D8 Advance diffractometer (Bruker GmbH, Karlsruhe, Germany) in a theta-theta reflection mode with copper K-α radiation. Each sample was scanned in the 2θ range from 2° to 55° with the step size of 0.02° of 2θ. The data collection and interpretations were performed using DiffracPlus and the EVA V.16 programme, respectively.
The nitrogen adsorption and desorption isotherms were obtained at a relative pressure (P/P0) range of 0.05 to 1 on a Gemini 2380 instrument (Micromeritics Instrument Corporation, UK) for both free and drug-impregnated silica particles. The samples (100–150 mg) were degassed with nitrogen at 313.2 K for ~ 12 h prior to the measurements. The specific surface area (SSA) was calculated using the multipoint Brunauer–Emmett–Teller (BET) model whilst the total pore volume and distribution were estimated by the Barrett, Joyner, and Helena (BJH) model [87, 88].
In vitro Dissolution Studies
The desorption rate of PRG and TST from drug-loaded silica was evaluated in pH 6.8 phosphate buffer solution (PBS) containing 1% sodium dodecyl sulphate (SDS) at 310.2 K. SDS is an anionic surfactant that is commonly added in the media at concentrations above its critical micellisation concentration (CMC) to overcome the sink limitations when determining the dissolution of hydrophobic drugs or formulations containing such molecules [89]. The dissolution of unprocessed TST and PRG in the release media was also investigated to allow for comparative data. Accordingly, 50 mg of drug-loaded ExP particles or the equivalent amount of pure PRG/TST was suspended in 15 mL of desorption media and stirred at 100 rpm. At regular time intervals of 2, 5, 10, 15, 30, 45, 60, 90, 120, 180, and 240 min, 3 mL of the release media was removed and replaced with the same volume of fresh PBS. Samples were then filtered and analysed by UV–Vis spectroscopy at 240 nm to quantify the drug release. The drug release experiments were carried out in triplicate and the percentage drug content in the media at each time point was calculated using Eq. (1):
where D%: drug release (%), Cn: corrected concentration (mg/mL), VPBS: volume of dissolution media (mL), and the “Impregnated mass of drug” relates to the total theoretical amount of drug in the sample.
Results and Discussion
scCO2 Impregnation of PRG and TST on Silica Particles
Both PRG and TST impregnation on silica were performed in scCO2 under various pressures and temperatures to determine the best parameters for drug loading.
Effect of scCO2 Processing Parameters on Drug Loading
The use of porous silica as carriers for therapeutic molecules is relatively common especially amorphous silica as used in this study because of its reduced toxicity in comparison to the crystalline form [42]. The effectiveness of the scCO2 technique for the impregnation of PRG and TST on mesoporous silica particles was investigated by varying the working pressure, temperature, and equilibration time. The data collected for PRG adsorption studies is presented in Table II and it indicates that there was mainly an effect of pressure and time on the amount of drug loaded onto silica.
At a constant time and temperature, an increase in pressure from 10 to 16 MPa led to an increase in PRG loading. However, this enhancement was either limited or absent when the pressure was increased from 16 to 18 MPa (i.e. an increase in pressure from 16 to 18 MPa led to either no change or a reduction in the PRG adsorption at a fixed temperature and time). Generally, at a specific pressure and time, the increase in temperature from 308 to 328 K resulted in reduced drug adsorption. This reduction in PRG loading was greater at 10 MPa than for the experiments conducted at higher pressures (16 and 18 MPa). At isobaric and isothermal conditions, a longer equilibration time always resulted in a significantly higher PRG loading. Further experiments were conducted using high pore size silica to determine if that leads to an increase in PRG adsorption at 16 and 18 MPa, and 308.2 and 328.2 K. The increase in pore size had an inverse effect on the drug loading as evident from Table II where PRG adsorption ranged from 6.4 to 9.2 mg/g. It is believed that there is a threshold value of pore size for the complete filling of the channels [90]. Pore sizes lower or higher than the threshold either restrict the diffusion of drug molecules or result in an underutilization of the total pore volume [91]. 9396 silica particles contain larger pore sizes than ExP which may lead to incomplete packing inside the pores due to already-loaded PRG molecules and restrict further pore occupation. Hence, further experiments were performed using only ExP particles.
The effect of time on PRG loading was further studied with experiments at 18 MPa and 328 K (Table III). The drug loading on silica improved significantly (i.e. from 5 to 82 mg/g) when the equilibration time was increased from 30 to 360 min.
Similarly, the findings reported for TST adsorption in Table IV show a greater effect of pressure and time on drug loading.
There was a rise in the TST loading on silica with the increase in pressure and time, whilst the rise in temperature from 308 to 328 K led to lower TST adsorption. Overall, the PRG loading onto ExP was significantly higher than that for TST.
The differences observed in the TST and PRG loading on silica and the effect of the pressure and temperature can be linked to disparities in drug solubility at a given condition in the scCO2. Kosal et al. reported solubility of PRG and TST in scCO2 at pressures of 8.1 to 25.3 MPa and temperatures between 328 and 333 K [85]. Their findings showed that the solubility of PRG is higher in scCO2 than TST and the solubility of both improves as the pressure or the solvent density increases. On the other hand, raising the temperature leads to a decrease in PRG/TST solubility due to the reduction in the density and solvation power of CO2. Thus, it could be theorised that the adsorption of PRG/TST on silica observed in this study was solubility limited. In this work, the increase in pressure from 10 to 18 MPa at constant temperature favoured drug solubility, resulting in the subsequently increased adsorption of both drugs on silica. The improvement in drug adsorption with increased equilibration time indicated that a minimum contact time is required for optimum loading efficiency. Further experiments for TST adsorption were not conducted as its adsorption was found to be extremely limited in comparison. Experiments with a co-solvent were assumed to be a better strategy to improve TST loading which is discussed in the following section.
Effect of Co-solvent on Drug Loading
The influence of co-solvent on the scCO2 adsorption of PRG and TST onto silica was studied at pressures of 16 MPa and 18 MPa and at temperatures of 308 and 328 K in the presence of THF for a contact time of 120 min. These co-solvent experiments were omitted at 100 bar due to the comparatively limited drug loading at this pressure. THF was selected as an appropriate co-solvent as it is an excellent solvent for both TST and PRG when compared to other common organic solvents such as acetone and ethanol [92].
As noted in Table V, there was a general increase in PRG loading with the addition of co-solvent aside from the study conducted at 16 MPa and 328 K where there was no change to the drug impregnation. At the contact time of 120 min, the PRG adsorption increased from 24 mg/g (without co-solvent) to 53 mg/g with the addition of THF. Similar to previous findings, a temperature increase had a negative impact on PRG impregnation at a given pressure. Surprisingly, the impact of pressure was not as apparent as the increase in pressure at 308 K resulted in the reduced drug loading, whereas there was only a slight increase in PRG impregnation at 328 K.
The impact of the addition of co-solvent on the TST impregnation on silica is presented in Table VI.
The increasing effect of co-solvent on drug loading was also observed for TST. As presented in Table VI, the absence of THF resulted in only 4 to 6 mg/g of TST impregnation, whereas the introduction of co-solvent improved the adsorption significantly resulting in the TST loading of 7 to 16 mg/g. Like the PRG adsorption studies, the TST impregnation was higher at a lower temperature and there was a reduction in the amount of TST adsorbed with the change in pressure from 160 to 180 bar. In general, the rise in TST and PRG loading observed with the use of co-solvent can be linked to the increase in drug solubility in the binary solvent system. The application of scCO2/co-solvent mixtures is known to improve drug solubility by altering the solvent density and increasing the solute–solvent interactions [93,94,95]. However, the increase in both the temperature and pressure can also result in diminishing the impact of co-solvent, possibly due to improved solvent–solvent interaction leading to the overall reduction in the solvent power of the binary mixture for a solute [96]. This may explain why an increase in pressure at both temperatures led to the low impregnation of both TST and PRG.
Characterisation
The presence of solvent residues in a pharmaceutical formulation may result in product instability and an increased risk of toxicity. Hence, it is imperative to assess if there are any remnants of organic solvents in cases where they are needed for product preparation. The retention times for ethanol and THF in GC were found to be at 3.6 and 5.2 min respectively. The calibration curve of THF was plotted in ethanol as presented as an inset in Fig. 2 and it was used to determine the residual solvent in formulations after processing. A representative GC chromatogram of PRG-adsorbed silica is presented in Fig. 2. The chromatograms collected for the formulations showed only the EtOH peak at 3.6 min, suggesting an absence of THF in the sample solution. THF is a class 2 solvent with a permitted daily allowance (PDE) of 7.2 mg/day [97]. The data obtained from GC suggests the absence of THF in the formulations or presence in extremely small quantities that is below the PDE limit.

Residual solvent determination (inset: THF calibration curve)
Based on the adsorption data, the PRG- and TST-loaded ExP particles were prepared in the presence of the co-solvent at 16 MPa and 308.2 K for 360 min. The drug loading for TST and PRG on silica at these processing conditions was approximately 16 ± 1.3 mg/g and 64 ± 1 mg/g, respectively.
DSC was performed to investigate the physical state of PRG and TST impregnated on silica after scCO2 processing. The thermograms for unprocessed and scCO2-processed drugs, and ExP, along with the drug-adsorbed formulations (PRG-ExP and TST-ExP), are presented in Fig. 3.

DSC thermograms for TST, PRG, ExP, TST-ExP, and PRG-ExP
The thermograms of unprocessed PRG and TST showed a sharp melting peak at approximately 406 and 424 K, respectively, due to their characteristic crystalline nature [92]. DSC analysis was also performed on PRG and TST processed at 308.2 K and 16 MPa. The thermograms presented in Fig. 3 showed similar onset of melting/melting peaks for both drugs. This indicates that CO2 processing had no effect on the thermal properties of TST and PRG, and both drugs retained their crystalline nature. The DSC thermogram of ExP displayed no peaks, confirming its amorphous nature. The disappearance of PRG and TST melt peaks on the DSC of drug-loaded particles indicated the amorphisation of the impregnated drug.
The ATR-FTIR spectra of ExP, PRG, TST, scCO2-processed drugs, and drug-loaded particles are presented in Fig. 4. As a result of the similarity in their structural composition, both PRG and TST do not show many differences in their ATR-FTIR spectra. Similarly, there were no differences in the ATR-FTIR spectra of scCO2-processed drugs and unprocessed TST/PRG. As presented in Fig. 4, the characteristic peaks corresponding to the stretching vibration of carbonyl groups appear at 1699 and 1661 cm−1 for PRG, and at 1656 cm−1 for TST. Also, the peak attributed to conjugated C = C stretching is observed at 1615 cm−1 (PRG) and 1612 cm−1 (TST) [98,99,100]. On the other hand, the ATR-FTIR spectra of PRG-ExP and TST-ExP overlap with that of unprocessed ExP particles, without displaying any significant peaks for the impregnated drugs probably due to proportionally higher silica content in the formulated systems. In general, the absorption bands near 1070 cm−1 relate to the stretching vibrations of Si–O bonds, whilst bands at 800 cm−1 are related to the bending vibrations of Si–O bonds. The bands near 450 cm−1 are attributed to bending vibrations of O-Si–O bonds [101, 102]. In general, vibrational spectra of amorphous materials (e.g. ExP particles) show broader and merged bands compared to crystalline solids owing to the lack of long-range order and the presence of various molecular conformations/intermolecular arrangements [103]. Although no bands related to TST and PRG could be seen between 400 and 2000 cm−1, band merging is evident in the CH-stretching region (2800 to 3100 cm−1) on the spectra of drug-loaded particles. The FTIR spectra of TST/PRG-loaded particles also suggest a weak interaction between the drug and silica, possibly via van der Waals and hydrogen bonding forces.

ATR-FTIR spectra for ExP, PRG, TST, and drug-loaded silica
The XRD analysis was performed to investigate possible changes in the crystalline structure of both scCO2-processed TST/PRG and the drug after loading on ExP particles. The diffractograms of PRG and TST (unprocessed and scCO2-processed), ExP, and drug-loaded particles are presented in Fig. 5. The major diffraction peaks of PRG and TST appeared in the 2θ range of 10.7 to 17°, therefore signifying the crystalline nature of both molecules [104, 105]. Moreover, diffractograms of processed and unprocessed drugs were identical confirming that the scCO2 processing in the absence of mesoporous silica did not cause any changes to the crystal structure of both actives. The XRD diffractograms obtained for free and drug-loaded silica showed no sharp peaks which is the characteristic of the amorphous form and confirm the absence of ordered crystalline structure [106].

XRD diffractograms of ExP, PRG, TST, and drug-loaded silica
The XRD data collected for PRG-ExP and TST-ExP particles confirm the findings observed with their DSC thermograms and indicate that the impregnated drugs are in the amorphous form, whereas scCO2 processing of TST and PRG in the absence of mesoporous silica does not cause any changes to their crystalline nature.
Nitrogen Adsorption Experiments
Nitrogen adsorption analyses were performed to determine changes in the surface properties of particles after drug impregnation. The BET isotherms in Fig. 6 showed the porous nature of both loaded and drug-free particles [107, 108].

BET isotherms for ExP, PRG-ExP, and TST-ExP (inset: BET isotherm from P/P0 values between 0.8 and 1)
The Type V isotherm for drug-free ExP remained unchanged after the impregnation of PRG and TST. The Type V isotherm is typically uncommon with the shape of the curve signifying weak interactions between the adsorbent (silica) and adsorbate (nitrogen) [107]. The specific surface area (SSA) calculated by the BET model along with the pore volume and diameter deduced by the BJH model for these silica particles is summarised in Table VII.
There was a decrease in the adsorbed nitrogen, SSA, and pore volume for drug-impregnated silica particles, when compared to the BET of ExP alone. This could be attributed to the reduced accessibility of adsorbing nitrogen molecules resulting from the presence of the drug on the surface and in the pores of silica particles. The recorded decrease in values (surface area and pore volume) was larger for PRG-ExP than for TST-ExP confirming favoured PRG impregnation using this method. This concurs with the adsorption data presented earlier where PRG adsorption at studied conditions was higher on ExP particles than for TST. It is common to see changes to the BET pore size and surface area of porous particles after adsorption, as this is indicative of the presence of the drug molecules in the pores. However, the degree of change is dependent on the quantity of drug adsorbed and the packing of pores by drug molecules (i.e. a higher adsorbed quantity may indicate high pore filling resulting in a greater difference and vice versa). The values reported in Table VII for TST-ExP were not significantly different to the silica particles itself which could once again be attributed to the low drug adsorption at the studied parameters. There was a slight increase in pore diameter for the drug-loaded particles when compared to the drug-free ExP. The BJH model is based on the cylindrical pore model and the pore diameter is calculated on the capillary condensation of the adsorbing nitrogen molecules in the pores. It does not consider any changes to the surface morphology which can be caused by the drug adsorption; hence, it can be unreliable in some cases; e.g. the BJH model can sometimes misinterpret adsorbate-associated surface imperfections as pores [109, 110]. Nevertheless, these results confirm the adsorption of PRG and TST on ExP as seen by the changes in SSA, pore volume, and nitrogen adsorption of particles after drug adsorption.
In vitro Dissolution Studies
The dissolution profiles of unprocessed and scCO2-processed PRG and TST, as well as for the drug-loaded particles (PRG-ExP and TST-ExP), are presented in Fig. 7.

Release kinetics from drug-loaded ExP, TST, PRG, and scCO2-treated TST and PRG at 310.2 K in PBS (inset: dissolution between 0 and 15 min)
As shown in the kinetics data, the dissolution of impregnated PRG and TST was higher than that of the corresponding unprocessed and scCO2-processed drugs. The dissolution occurred rapidly within the first 15 min and then remained relatively constant for the rest of the study, alluding to the improved solubility of both TST and PRG due to the adsorption on mesoporous silica. The drug dissolution from ExP appears to follow a first-order (R2: 0.9665) kinetics suggesting a diffusion-controlled release of both drugs from silica particles [111]. The drugs on their own had an expected slower and lower dissolution over the same timeframe. The dissolution of both processed and scCO2-processed drugs followed a similar trend, where it gradually increased until a plateau was achieved at 90 min with approximately 50–55% of the drug getting into the media. As mentioned earlier, the release of impregnated drugs was fast with ~ 80–90% obtained for both TST and PRG in the first 2 min in comparison to only 25% of unimpregnated actives. The high dissolution rate of drug–silica preparations can be attributed to the drug amorphisation along with the improved wettability [11, 112, 113]. An increase in the surface area due to the distribution of drugs in the micropores as well as favourable wettability permits accelerated penetration of the aqueous media into the capillaries on the porous particles resulting in the faster dissolution of both TST and PRG.
Conclusions
The impregnation of PRG and TST on silica particles was successfully achieved via a scCO2 process, where processing temperature, pressure, time, and co-solvent and their impact on the efficiency of drug impregnation were studied. The processing parameters had a significant impact on drug adsorption that ranged from 1 to 82 mg/g for PRG and 0.1 to 16 mg/g for TST. In general, changes in processing pressure and time had a considerable impact on drug adsorption, and the incorporation of PRG and TST onto silica particles was greatly improved with a rise in time and pressure. Also, the use of THF as a co-solvent led to a further increase in drug impregnation. Whereas the impact of temperature was comparatively limited, an increase in temperature either led to no change or a decrease in drug adsorption. The drug loading on mesoporous silica for PRG was higher than for TST which can be attributed to the higher solubility of PRG in scCO2. The presence of TST and PRG at the silica surface was confirmed by nitrogen adsorption experiments and the amorphous nature of the drug was verified with DSC and XRD analysis. The adsorption of PRG and TST onto silica led to an improvement in their solubility in aqueous media with approximately 90% of the drug dissolving in the first 2 min in comparison to only 25% of the free drug.
References
Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50:47–60.
Vo C, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85:799–813.
Subongkot T, Ngawhirunpat T. Development of a novel microemulsion for oral absorption enhancement of all-trans retinoic acid. Int J Nanomedicine. 2017;12:5585–99.
Kumar M, Bishnoi RS, Shukla AK, Jain CP. Techniques for formulation of nanoemulsion drug delivery system: a review. Prev Nutr Food Sci. 2019;24:225–34.
Lavasanifar A, Samuel J, Kwon GS. Poly(ethylene oxide)-block-poly(L-amino acid) micelles for drug delivery. Adv Drug Deliv Rev. 2002;54:169–90.
Carneiro SB, Duarte FÍC, Heimfarth L, Quintans JDSS, Quintans-Júnior LJ, Júnior VFDV, et al. Cyclodextrin-drug inclusion complexes: in vivo and in vitro approaches. Int J Mol Sci. 2019;20:642.
Kumar R, Thakur AK, Chaudhari P, Banerjee N. Particle size reduction techniques of pharmaceutical compounds for the enhancement of their dissolution rate and bioavailability. J Pharm Innov. 2022;17:333–52.
Babu NJ, Nangia A. Solubility advantage of amorphous drugs and pharmaceutical cocrystals. Cryst Growth Des. 2011;11:2662–79.
Rautio J, Meanwell NA, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17:559–87.
Mukherjee S, Ray S, Thakur RS. Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci. 2009;71:349.
Sanganwar GP, Gupta RB. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int J Pharm. 2008;360:213–18.
Ahern RJ, Hanrahan JP, Tobin JM, Ryan KB, Crean AM. Comparison of fenofibrate-mesoporous silica drug-loading processes for enhanced drug delivery. European Journal of Pharmaceutical Sciences. 2013;50:400–9.
Horcajada P, Rámila A, Pérez-Pariente J, Vallet-Regí M. Influence of pore size of MCM-41 matrices on drug delivery rate. Microporous and Mesoporous Materials. 2004;68:105–9.
Salonen J, Laitinen L, Kaukonen AM, Tuura J, Björkqvist M, Heikkilä T, et al. Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs. Journal of Controlled Release. 2005;108:362–74.
Lin YS, Hurley KR, Haynes CL. Critical considerations in the biomedical use of mesoporous silica nanoparticles. J Phys Chem Lett. 2012;3:364–74.
Girija AR, Balasubramanian S. Theragnostic potentials of core/shell mesoporous silica nanostructures. Nanotheranostics. 2019;3:1-40.
Frickenstein AN, Hagood JM, Britten CN, Abbott BS, McNally MW, Vopat CA, et al. Mesoporous silica nanoparticles: properties and strategies for enhancing clinical effect. Pharmaceutics. 2021;13:570.
Huang PK, Lin SX, Tsai MJ, Leong MK, Lin SR, Kankala RK, et al. Encapsulation of 16-hydroxycleroda-3,13-dine-16,15-olide in mesoporous silica nanoparticles as a natural dipeptidyl peptidase-4 inhibitor potentiated hypoglycemia in diabetic mice. Nanomaterials. 2017;7:112.
Tan L, Yang MY, Wu HX, Tang ZW, Xiao JY, Liu CJ, et al. Glucose- and pH-responsive nanogated ensemble based on polymeric network capped mesoporous silica. ACS Appl Mater Interfaces. 2015;7:6310–16.
Murugan C, Rayappan K, Thangam R, Bhanumathi R, Shanthi K, Vivek R, et al. Combinatorial nanocarrier based drug delivery approach for amalgamation of anti-tumor agents in breast cancer cells: an improved nanomedicine strategies. Sci Rep. 2016;6:34053.
Fu Q, Hargrove D, Lu X. Improving paclitaxel pharmacokinetics by using tumor-specific mesoporous silica nanoparticles with intraperitoneal delivery. Nanomedicine. 2016;12:1951–59.
di Pasqua AJ, Yuan H, Chung Y, Kim JK, Huckle JE, Li C, et al. Neutron-activatable holmium-containing mesoporous silica nanoparticles as a potential radionuclide therapeutic agent for ovarian cancer. J Nucl Med. 2013;54:111–6.
Alexa IF, Ignat M, Popovici RF, Timpu D, Popovici E. In vitro controlled release of antihypertensive drugs intercalated into unmodified SBA-15 and MgO modified SBA-15 matrices. Int J Pharm Elsevier. 2012;436:111–9.
Ganesh M, Ubaidulla U, Hemalatha P, Peng MM, Jang HT. Development of duloxetine hydrochloride loaded mesoporous silica nanoparticles: characterizations and in vitro evaluation. AAPS PharmSciTech. 2015;16:644–951.
Koneru B, Shi Y, Wang YC, Chavala SH, Miller ML, Holbert B, et al. Tetracycline-containing MCM-41 mesoporous silica nanoparticles for the treatment of Escherichia coli. Molecules. 2015;20:19690–98.
Zhou X, Feng W, Qiu K, Chen L, Wang W, Nie W, et al. BMP-2 derived peptide and dexamethasone incorporated mesoporous silica nanoparticles for enhanced osteogenic differentiation of bone mesenchymal stem cells. ACS Appl Mater Interfaces. 2015;7:15777–89.
Tautzenberger A, Kovtun A, Ignatius A. Nanoparticles and their potential for application in bone. Int J Nanomedicine. 2012:7:4545.
He Q, Shi J. Mesoporous silica nanoparticle based nano drug delivery systems: synthesis, controlled drug release and delivery, pharmacokinetics and biocompatibility. J Mater Chem. 2011;21:5845.
Tan A, Eskandar NG, Rao S, Prestidge CA. First in man bioavailability and tolerability studies of a silica-lipid hybrid (Lipoceramic) formulation: a phase I study with ibuprofen. Drug Deliv Transl Res. 2014;4:212–21.
Bukara K, Schueller L, Rosier J, Martens MA, Daems T, Verheyden L, et al. Ordered mesoporous silica to enhance the bioavailability of poorly water-soluble drugs: proof of concept in man. Eur J Pharm Biopharm. 2016;108:220–5 (Elsevier).
Phillips E, Penate-Medina O, Zanzonico PB, Carvajal RD, Mohan P, Ye Y, et al. Clinical translation of an ultrasmall inorganic optical-PET imaging nanoparticle probe. Sci Transl Med. 2014;6:260ra149.
Janjua TI, Cao Y, Yu C, Popat A. Clinical translation of silica nanoparticles. Nat Rev Mater. 2021:6:1072–74.
Niculescu VC. Mesoporous silica nanoparticles for bio-applications. Front Mater. 2020:6: https://doi.org/10.3389/fmats.2020.00036.
Song SW, Hidajat K, Kawi S. Functionalized SBA-15 materials as carriers for controlled drug delivery: influence of surface properties on matrix-drug interactions. Langmuir. 2005;21:9568–75.
Bouledjouidja A, Masmoudi Y, van Speybroeck M, Schueller L, Badens E. Impregnation of fenofibrate on mesoporous silica using supercritical carbon dioxide. Int J Pharm. 2016;499:1–9.
Qian KK, Bogner RH. Spontaneous crystalline-to-amorphous phase transformation of organic or medicinal compounds in the presence of porous media, part 1: thermodynamics of spontaneous amorphization. J Pharm Sci. 2011;100:2801–15.
Mellaerts R, Jammaer JAG, van Speybroeck M, Hong C, van Humbeeck J, Augustijns P, et al. Physical state of poorly water soluble therapeutic molecules loaded into SBA-15 ordered mesoporous silica carriers: a case study with itraconazole and ibuprofen. Langmuir. 2008;24:8651–59.
Charnay C, Bégu S, Tourné-Péteilh C, Nicole L, Lerner DA, Devoisselle JM. Inclusion of ibuprofen in mesoporous templated silica: drug loading and release property. Eur J Pharm Biopharm. 2004;57:533–40.
van Speybroeck M, Barillaro V, Thi T do, Mellaerts R, Martens J, van Humbeeck J, et al. Ordered mesoporous silica material SBA-15: a broad-spectrum formulation platform for poorly soluble drugs. J Pharm Sci. 2009;98:2648–58.
Mellaerts R, Mols R, Jammaer JAG, Aerts CA, Annaert P, van Humbeeck J, et al. Increasing the oral bioavailability of the poorly water soluble drug itraconazole with ordered mesoporous silica. Eur J Pharm Biopharm. 2008;69:223–30.
Andersson J, Rosenholm J, Areva S, Lindén M. Influences of material characteristics on ibuprofen drug loading and release profiles from ordered micro- and mesoporous silica matrices. Chem Mater. 2004;16:4160–67.
Trzeciak K, Chotera‐ouda A, Bak‐sypien II, Potrzebowski MJ. Mesoporous silica particles as drug delivery systems—the state of the art in loading methods and the recent progress in analytical techniques for monitoring these processes. Pharmaceutics. 2021;13:950.
Seljak KB, Kocbek P, Gašperlin M. Mesoporous silica nanoparticles as delivery carriers: an overview of drug loading techniques. J Drug Deliv Sci Technol. 2020;59:101906 (Elsevier).
Nandi U, Ajiboye AL, Patel P, Douroumis D, Trivedi V. Preparation of solid dispersions of simvastatin and Soluplus using a single-step organic solvent-free supercritical fluid process for the drug solubility and dissolution rate enhancement. Pharmaceuticals. 2021;14:846.
Fages J, Lochard H, Letourneau JJ, Sauceau M, Rodier E. Particle generation for pharmaceutical applications using supercritical fluid technology. Powder Technol. 2004;141:219–26.
Bhomia R, Trivedi V, Mitchell JC, Coleman NJ, Snowden MJ. Effect of pressure on the melting point of pluronics in pressurized carbon dioxide. Ind Eng Chem Res. 2014;53:10820–25.
Duarte ARC, Mano JF, Reis RL. Supercritical fluids in biomedical and tissue engineering applications: a review. Int Mater Rev. 2009;54:214–22.
Knez Z, Weidner E. Particles formation and particle design using supercritical fluids. Curr Opin Solid State Mater Sci. 2003;7:353–61.
Kalani M, Yunus R. Effect of supercritical fluid density on nanoencapsulated drug particle size using the supercritical antisolvent method. Int J Nanomedicine. 2012;7:2165.
Kalani M, Yunus R. Application of supercritical antisolvent method in drug encapsulation: a review. Int J Nanomedicine. 2011;6:1429.
Kemmere MF. Supercritical carbon dioxide for sustainable polymer processes. Supercrit Carbon Dioxide: in Polymer Reaction Engineering. 2006;1:1–14.
Belhadj-Ahmed F, Badens E, Llewellyn P, Denoyel R, Charbit G. Impregnation of vitamin E acetate on silica mesoporous phases using supercritical carbon dioxide. J Supercrit Fluids. 2009;51:278–86.
Smirnova I, Suttiruengwong S, Seiler M, Arlt W. Dissolution rate enhancement by adsorption of poorly soluble drugs on hydrophilic silica aerogels. Pharm Dev Technol. 2004;9:443–52.
García-Casas I, Crampon C, Montes A, Pereyra C, Martínez de la Ossa EJ, Badens E. Supercritical CO2 impregnation of silica microparticles with quercetin. Journal of Supercritical Fluids. 2019;143:157–61.
Smirnova I, Suttiruengwong S, Arlt W. Feasibility study of hydrophilic and hydrophobic silica aerogels as drug delivery systems. J Non Cryst Solids. 2004;350:54–60.
Wang LH, Che X, Xu H, Zhou LL, Han J, Zou MJ, et al. A novel strategy to design sustained-release poorly water-soluble drug mesoporous silica microparticles based on supercritical fluid technique. Int J Pharm. 2013;454:135–42.
García-Casas I, Montes A, Valor D, Pereyra C, Martínez de la Ossa EJ. Impregnation of mesoporous silica with mangiferin using supercritical CO2. J Supercrit Fluids. 2018;140:139–6.
Singh N, Vinjamur M, Mukhopadhyay M. Influence of drug properties on loadings and release kinetics of drugs from silica aerogels loaded in supercritical CO2. J Supercrit Fluids. 2022;181:105510.
Morre J, Tenorio MJ, Torralvo MJ, Pando C, Renuncio JAR, Cabañas A. Deposition of Pd into mesoporous silica SBA-15 using supercritical carbon dioxide. J Supercrit Fluids. 2011;56:213–22.
Gignone A, Manna L, Ronchetti S, Banchero M, Onida B. Incorporation of clotrimazole in ordered mesoporous silica by supercritical CO2. Microporous Mesoporous Mater. 2014;200:291–6.
Jiang Q, Li Y, Fu Q, Geng Y, Zhao J, Ma P, et al. In-vitro and in-vivo study of amorphous spironolactone prepared by adsorption method using supercritical CO2. Drug Dev Ind Pharm. 2015;41:201–6.
Smirnova I, Suttiruengwong S, Arlt W. Aerogels: tailor-made carriers for immediate and prolonged drug release. KONA Powder and Particle Journal. 2005;23:86–97.
Yang D, Jiang Q, Ma J, Guo B, Li Y, Ma P, et al. Nisoldipine dissolution profile enhancement by supercritical carbon dioxide impregnation technique with fumed silica. Powder Technol. 2015;271:7–15.
Yang G, Li Z, Wu F, Chen M, Wang R, Zhu H, et al. Improving solubility and bioavailability of breviscapine with mesoporous silica nanoparticles prepared using ultrasound-assisted solution-enhanced dispersion by supercritical fluids method. Int J Nanomedicine. 2020;15:1661–75.
Raza A, Alavi SE, Sime FB, Han FY, Roberts JA, Popat A, et al. Microfluidic assembly of pomegranate-like hierarchical microspheres for efflux regulation in oral drug delivery. Acta Biomater. 2021;126:277–90
Raza A, Sime FB, Cabot PJ, Roberts JA, Falconer JR, Kumeria T, et al. Liquid CO2 formulated mesoporous silica nanoparticles for pH-responsive oral delivery of meropenem. ACS Biomater Sci Eng. 2021;7:1836–53.
Gallo M, Serpella L, Leone F, Manna L, Banchero M, Ronchetti S, et al. Piroxicam loading onto mesoporous silicas by supercritical CO2 impregnation. Molecules. 2021;26:2500.
Hussain AA, Al-Bayatti AA, Dakkuri A, Okochi K, Hussain MA. Testosterone 17β-N,N-dimethylglycinate hydrochloride: a prodrug with a potential for nasal delivery of testosterone. J Pharm Sci. 2002;91:785–9.
Vinarov Z, Dobreva P, Tcholakova S. Effect of surfactant molecular structure on progesterone solubilization. J Drug Deliv Sci Technol. 2018;43:44–9 (Elsevier).
Savla R, Browne J, Plassat V, Wasan KM, Wasan EK. Review and analysis of FDA approved drugs using lipid-based formulations. Drug Dev Ind Pharm. 2017;43:1743–58.
Yunxia Y, Huihui N, Shiying X, yingwa G, Xiangxiang W. Solubility and dissolution rate of progesterone cocrystals using 4-fluorobenzoic acid and 2-hydroxy-6-naphthoic acid as coformers. J Cryst Growth. 2022;585:126601.
Shankar VK, Police A, Pandey P, Cuny ZG, Repka MA, Doerksen RJ, et al. Optimization of sulfobutyl-ether-β-cyclodextrin levels in oral formulations to enhance progesterone bioavailability. Int J Pharm. 2021;596:120212.
Lahiani-Skiba M, Barbot C, Bounoure F, Joudieh S, Skiba M. Solubility and dissolution rate of progesterone-cyclodextrin-polymer systems. Drug Dev Ind Pharm. 2006;32:1043–58.
Celia C, Scala A, Stancanelli R, Surdo E, Paolino D, Grattoni A, et al. Physicochemical properties of inclusion complexes of highly soluble β-cyclodextrins with highly hydrophobic testosterone propionate. Int J Pharm. 2017;534:316–24.
Pitha J, Harman SM, Michel ME. Hydrophilic cyclodextrin derivatives enable effective oral administration of steroidal hormones. J Pharm Sci. 1986;75:165–7.
Pitha J, Pitha J. Amorphous water‐soluble derivatives of cyclodextrins: nontoxic dissolution enhancing excipients. J Pharm Sci. 1985;74:987–90.
Guilherme MR, Mauricio MR, Tenório-Neto ET, Kunita MH, Cardozo-Filho L, Cellet TSP, et al. Polycaprolactone nanoparticles containing encapsulated progesterone prepared using a scCO2 emulsion drying technique. Mater Lett. 2014;124:197–200.
Fogolari O, Felippe AC, Leimann FV, Gonçalves OH, Sayer C, de Araujo PHH. Method validation for progesterone determination in poly(methyl methacrylate) nanoparticles synthesized via miniemulsion polymerization. Int J Polym Sci. 2017;2017:1–11.
Pasqua L, Veltri L, Gabriele B, Testa F, Salerno G. Progesterone inclusion into cyclodextrin-functionalized mesoporous silica. J Porous Mater. 2013;20:917–25.
Giri K, Kuschnerus I, Ruan J, Garcia-Bennett AE. Influence of a protein corona on the oral pharmacokinetics of testosterone released from mesoporous silica. Adv Ther (Weinh). 2020;3:1900110.
Gordon J, Kazemian H, Rohani S. MIL-53(Fe), MIL-101, and SBA-15 porous materials: potential platforms for drug delivery. Mater Sci Eng C. 2015;47:172–9.
Huang Z, Guo Y hua, Miao H, Teng L jun. Solubility of progesterone in supercritical carbon dioxide and its micronization through RESS. Powder Technol. 2014;258:66–77.
Irena Z. Potential of supercritical solvent impregnation for development of materials with antibacterial properties. Int Arch Med Microbiol. 2017;1. https://doi.org/10.23937/iamm-2017/1710001.
Alessi P, Cortesi A, Kikic I, Foster NR, Macnaughton SJ, Colombo I. Particle production of steroid drugs using supercritical fluid processing. Ind Eng Chem Res. 1996;35:4718–26.
Kosal E, Lee CH, Holder GD. Solubility of progesterone, testosterone, and cholesterol in supercritical fluids. J Supercrit Fluids. 1992;5:169–79.
Rudrangi SRS, Bhomia R, Trivedi V, Vine GJ, Mitchell JC, Alexander BD, et al. Influence of the preparation method on the physicochemical properties of indomethacin and methyl-β-cyclodextrin complexes. Int J Pharm. 2015;479:318–90.
Brunauer S, Emmett PH, Teller E. Adsorption of gases in multimolecular layers. J Am Chem Soc. 1938;60:309–19.
Huang X, Young NP, Townley HE. Characterization and comparison of mesoporous silica particles for optimized drug delivery. Nanomater Nanotechnol. 2014;4. https://doi.org/10.5772/58290.
Phillips DJ, Pygall SR, Cooper VB, Mann JC. Overcoming sink limitations in dissolution testing: a review of traditional methods and the potential utility of biphasic systems. J Pharm Pharmacol. 2012;64:1549–59.
Horcajada P, Rámila A, Pérez-Pariente J, Vallet-Regı́ M. Influence of pore size of MCM-41 matrices on drug delivery rate. Microporous and Mesoporous Materials. 2004;68:105–9.
Li J, Shen S, Kong F, Jiang T, Tang C, Yin C. Effects of pore size on: in vitro and in vivo anticancer efficacies of mesoporous silica nanoparticles. RSC Adv. 2018;8:24633–40.
Shen W, Sun W, Yang W, Xu H, Hu G, Zhao G, et al. Solubility determination and thermodynamic modelling of progesterone in twelve pure solvents and three binary solvents at 278.15 to 323.15 K. J Mol Liq. 2021;324:114715.
Kopcak U, Mohamed RS. Caffeine solubility in supercritical carbon dioxide/co-solvent mixtures. J Supercrit Fluids. 2005;34:209–14.
Sauceau M, Letourneau JJ, Freiss B, Richon D, Fages J. Solubility of eflucimibe in supercritical carbon dioxide with or without a co-solvent. J Supercrit Fluids. 2004;31:133–40.
Ting SST, Macnaughton SJ, Tomasko DL, Foster NR. Solubility of naproxen in supercritical carbon dioxide with and without cosolvents. Ind Eng Chem Res. 1993;32:1471–81.
Behjati Rad H, Karimi Sabet J, Varaminian F. Effect of stearic acid as a co-solvent on the solubility enhancement of aspirin in supercritical CO2. Chem Eng Technol. 2019;42:1259–67.
Parris P, Duncan JN, Fleetwood A, Beierschmitt WP. Calculation of a permitted daily exposure value for the solvent 2-methyltetrahydrofuran. Regul Toxicol Pharmacol. 2017;87:54–63.
Leimann FV, Biz MH, Kaufmann KC, Maia WJ, Honçalves OH, Filho LC, et al. Characterization of progesterone loaded biodegradable blend polymeric nanoparticles. Ciencia Rural. 2015;45:2082–88.
Chen X, Partheniadis I, Nikolakakis I, Al-Obaidi H. Solubility improvement of progesterone from solid dispersions prepared by solvent evaporation and co-milling. Polymers (Basel). 2020;12:854.
Tajbakhsh M, Saeedi M, Akbari J, Morteza-Semnani K, Nokhodchi A, Hedayatizadeh-Omran A. An investigation on parameters affecting the optimization of testosterone enanthate loaded solid nanoparticles for enhanced transdermal delivery. Colloids Surf A Physicochem Eng Asp. 2020;589:124437.
Wang Z, Liu Q, Yu J, Wu T, Wang G. Surface structure and catalytic behavior of silica-supported copper catalysts prepared by impregnation and sol-gel methods. Appl Catal A Gen. 2003;239:87–94.
Khdary NH, Ghanem MA, Abdesalam ME, Al-Garadah MM. Sequestration of CO2 using Cu nanoparticles supported on spherical and rod-shape mesoporous silica. J Saudi Chem Soc. 2018;22:343–51.
Planinšek O, Kovačič B, Vrečer F. Carvedilol dissolution improvement by preparation of solid dispersions with porous silica. Int J Pharm. 2011;406:41–8.
Falconer JR, Wen J, Zargar-Shoshtari S, Chen JJ, Farid M, Tallon SJ, et al. Preparation and characterization of progesterone dispersions using supercritical carbon dioxide. Drug Dev Ind Pharm. 2014;40:458–69.
Muchow M, Maincent P, Mller RH, Keck CM. Production and characterization of testosterone undecanoate-loaded NLC for oral bioavailability enhancement. Drug Dev Ind Pharm. 2011;37:8–14.
Nallathambi G, Ramachandran T, Rajendran V, Palanivelu R. Effect of silica nanoparticles and BTCA on physical properties of cotton fabrics. Mater Res. 2011;14:552–9.
Sing KSW, Everett DH, Haul RAW, Moscou L, Pierotti RA, Rouquerol J, et al. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity. Pure Appl Chem. 1985;57:603–19.
Alothman ZA. A review: fundamental aspects of silicate mesoporous materials. Materials. 2012;5:2874–902.
Horikawa T, Do DD, Nicholson D. Capillary condensation of adsorbates in porous materials. Adv Colloid Interface Sci. 2011;169:40–58.
de Lange MF, Vlugt TJH, Gascon J, Kapteijn F. Adsorptive characterization of porous solids: error analysis guides the way. Microporous Mesoporous Mater. 2014;200:199–215.
Talevi A, Ruiz ME. Drug Release 2021 The ADME encyclopedia: a comprehensive guide on biopharmacy and pharmacokinetics [Internet]. Cham: Springer International Publishing; 2021. p. 1–7. https://doi.org/10.1007/978-3-030-51519-5_32-1.
Wang X, Zhang L, Ma D, Tang X, Zhang Y, Yin T, et al. Characterizing and exploring the differences in dissolution and stability between crystalline solid dispersion and amorphous solid dispersion. AAPS PharmSciTech. 2020;21:262.
Teja SB, Patil SP, Shete G, Patel S, Bansal AK. Drug-excipient behavior in polymeric amorphous solid dispersions. J Excip Food Chem. 2014;4:70–94.
Funding
This study was funded by the University of Kent.
Author information
Authors and Affiliations
Contributions
Conceptualization: Vivek Trivedi, Claudia Mattern, Uttom Nandi, and Adejumoke Lara Ajiboye. Investigation: Amélie Jacopin, Uttom Nandi, Andrew Hurt, and Adejumoke Lara Ajiboye. Resources: Vivek Trivedi and Claudia Mattern. Writing—original draft preparation: Amélie Jacopin, Adejumoke Lara Ajiboye, Uttom Nandi, and Vivek Trivedi. Writing—review and editing: Adejumoke Lara Ajiboye, Vivek Trivedi, and Claudia Mattern. Supervision: Vivek Trivedi and Claudia Mattern. Project administration: Vivek Trivedi and Adejumoke Lara Ajiboye.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ajiboye, A.L., Jacopin, A., Mattern, C. et al. Dissolution Improvement of Progesterone and Testosterone via Impregnation on Mesoporous Silica Using Supercritical Carbon Dioxide. AAPS PharmSciTech 23, 302 (2022). https://doi.org/10.1208/s12249-022-02453-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-022-02453-z