
Abstract
G protein-coupled receptors (GPCRs) have important functions in both innate and adaptive immunity, with the capacity to bridge interactions between the two arms of the host responses to pathogens through direct recognition of secreted microbial products or the by-products of host cells damaged by pathogen exposure. In the mid-1990s, a large group of intracellular proteins was discovered, the regulator of G protein signaling (RGS) family, whose main, but not exclusive, function appears to be to constrain the intensity and duration of GPCR signaling. The R4/B subfamily—the focus of this review—includes RGS1–5, 8, 13, 16, 18, and 21, which are the smallest RGS proteins in size, with the exception of RGS3. Prominent roles in the trafficking of B and T lymphocytes and macrophages have been described for RGS1, RGS13, and RGS16, while RGS18 appears to control platelet and osteoclast functions. Additional G protein independent functions of RGS13 have been uncovered in gene expression in B lymphocytes and mast cell-mediated allergic reactions. In this review, we discuss potential physiological roles of this RGS protein subfamily, primarily in leukocytes having central roles in immune and inflammatory responses. We also discuss approaches to target RGS proteins therapeutically, which represents a virtually untapped strategy to combat exaggerated immune responses leading to inflammation.
Similar content being viewed by others
INTRODUCTION
The Pleotropic Roles of GPCR Signaling in Immune Responses
Innate immune host defenses are activated through recognition of microorganism-associated molecular patterns (MAMPs), which then stimulate pattern recognition receptors (PRRs) including Toll-like, nucleotide-binding oligomerization domain (NOD), and C-type lectin receptors on professional phagocytic cells including macrophages, dendritic cells, and granulocytes (1). If invading microbes cannot be readily and immediately eliminated through these and other mechanisms, antigen-presenting cells (APCs) including dendritic cells, macrophages, and possibly basophils process and display foreign antigens to adaptive immune cells in lymphoid organs. These interactions lead to recognition by antigen-restricted adaptive immune cells (B and T lymphocytes) and subsequent eradication of pathogens through either cytotoxic T cell-mediated responses or the generation of neutralizing antibodies by B lymphocytes.
An ancient example of GPCR function in immune responses was recently uncovered in the nematode Caenorhabditis elegans. The endoparasitic fungus Drechmeria coniospora can systemically infect worms by invading the cuticle of the skin. This induces production of the tyrosine metabolite 4-hydroxyphenyllactic acid (HPLA) in the skin, which is recognized by GPCR DCA-1, triggering production of antimicrobial peptides. DCA-1 is required for systemic fungal resistance (2). In mammalian cells, similar recognition of microbial-derived substances including bacterial toxins, formyl peptides, and lipids by GPCRs occurs in both innate and adaptive immune cells. Many of these compounds act as chemoattractants for innate immune cells including monocyte/macrophages and granulocytes, eliciting their rapid recruitment to tissue portals of entry for invading microbes (3).
A special class of chemoattractant molecules, chemokines, are produced by host cells throughout the body and act on chemokine GPCRs expressed on both innate and adaptive immune leukocytes to elicit chemotaxis, i.e., directed migration. Chemokine gradients in the thymus and bone marrow drive thymocyte differentiation and exit from the thymus and egress of immature B lymphocytes into the bloodstream (4). In secondary lymphoid organs such as the spleen and lymph nodes (LNs), a precise combination of chemokines including CXCL12, 13, and 21 together with the serum lipid mediator sphingosine 1-phosphate (S1-P) direct B and T cell trafficking to and transit within these structures. These and other substances guide interactions with stromal cells (e.g., follicular dendritic cells) within the follicles, which are crucial for B lymphocyte differentiation and maturation and the generation of high-affinity antibody responses to foreign antigens. These processes and chemokines involved have been studied extensively and reviewed in detail elsewhere (5,6).
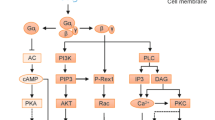
GPCRs signal through the heterotrimeric G protein complex of α, β, and γ subunits. Ligand activation of the GPCR induces exchange of GDP for GTP on the α subunit; Gα-GTP stimulates several classes of downstream effectors including kinases, phospholipases, and ion channels. GPCR activation elicits many cellular responses including chemotaxis, inflammatory gene transcription, and proliferation (7). The pathways activated by a particular GPCR are distinguished by the class of α subunit to which it couples: there are four subfamilies of α subunits: αi/o, αs, αq, and α12/13. Most chemokine receptors couple to Gαi, and chemokine-mediated cell migration is inhibited by pertussis toxin (PTX), which ADP-ribosylates of Gαi prevent receptor association (8). In contrast, other classes of chemoattractants—granulocyte formyl peptide receptors or lymphocyte phospholipid mediator receptors are two examples—couple to Gαq or Gα12/13, which are both resistant to inactivation by PTX (9).
Overexuberant chemoattractant signaling—or an imbalance between migration and retention signals in lymphoid organs and/or peripheral tissue—may result in inflammation, and in extreme instances autoimmune disease. The proinflammatory cytokine milieu associated with infections, trauma, or allergies may lead to increased leukocyte recruitment and retention at sites of inflammation due to altered expression and/or function of GPCRs or components of their downstream circuitry. GPCR desensitization to a continual stimulus provides a physiological brake on chemoattractant signaling, which is mediated by two principal mechanisms: (1) receptor phosphorylation by GPCR-associated kinases (GRKs) and (2) hydrolysis of GTP by the α subunit, which allows Gα-GDP to reassemble with Gβγ to form an inactive heterotrimer [(10,11) for recent reviews].
All members of the RGS superfamily, which number more than 30 in mammalian cells, contain a conserved RGS domain or “RGS box” (12,13). This region mediates RGS binding to activated Gα subunits and acceleration of GTP hydrolysis by Gα 10–1000-fold. RGS proteins can be further classified into subfamilies based on primary sequence homology and the presence of additional signaling domains (Fig. 1) (14). Members of the B/R4 subfamily of RGS proteins—consisting of RGS1–5, 8, 13, 16, 18, and 21—are of similar size (∼20–25 kDa) and are comprised of little more than the RGS box, with the exception of RGS3, which is larger (∼80 kDa) owing to its amino-terminal post synaptic density protein (PSD95), Drosophila disk large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1) [PDZ] domain. Nearly all of these RGS proteins act as GTPase accelerating proteins (GAP) for both Gαi and Gαq, but not Gαs or Gα12/13, except RGS2, which appears to be relatively specific for Gαq (15), and the A/RZ family, which may preferentially bind Gαz in addition to Gαi/o. Because of their structural simplicity, high degree of homology with each other, and enriched expression in the hematopoietic compartment (e.g., RGS1, (13,16,17)), we have focused on functions of R4 RGS proteins in the immune system in this review. Some but not all RGS activities in various tissues have been elucidated over the past 20 years (Table I). But their physiological functions remain poorly defined—there are no human diseases directly attributable to monogenic mutations and/or dysfunction of a specific RGS protein. Likewise, specific GPCR pathways in immune cells regulated by each R4 RGS protein have not been fully delineated. Functional redundancy of RGS activity is possible; highly restricted and context-dependent expression and regulation by protein modification and subcellular localization, for example, may override the basal expression of multiple RGS proteins in the same cell to specify precise regulation of select GPCR pathways (16).

Proteins containing a regulator of G protein signaling (RGS) domain. The various RGS subfamilies, together with their known members and distinguishing domain structures, are shown. Additional proteins containing RGS homology domains (lower right) are also included but are not discussed in this review. Abbreviations: PDZ post synaptic density protein (PSD95), Drosophila disk large tumor suppressor (Dlg1), and zonula occludens-1 protein (zo-1) [PDZ] domain, DEP Disheveled, Egl-10 and Pleckstrin domain, PTB protein tyrosine binding domain, RBD Ras binding domain, GGL Gγ-like domain
Biochemical and crystallography studies indicate that RGS GAP activity is mediated by direct binding to Gα proteins (17). Multiple residues of the RGS domain make contact with conserved amino acids in the so-called Switch regions in Gα (on account of their distinct conformations in GDP v. GTP-bound versions) that are critical for GTP hydrolysis. Among these, Thr182 in Gα Switch I of Gαi contacts several highly conserved residues in RGS proteins to form an inflexible transition state conformation that facilitates GTP hydrolysis (18). In studies of yeast, mutation of the nearby Gly184 in Gpa1 (Gα orthologue) to Ser was found to render the Gα subunit fully able to bind βγ, nucleotide, receptors, and effectors, yet be completely resistant to RGS binding and GAP activity (19). Subsequently, mice with a germline Gαi2 (G184S) mutation were generated, which have served as a particularly useful tool to probe the effects of global loss of RGS activities in specific tissues and organ systems (20–22).
G Protein Deficiency or RGS Resistance Leads to Immune Abnormalities
G proteins and RGS proteins also modulate B and T lymphocyte chemotaxis and entry and exit from the lymph nodes and thymus. Mature B and T lymphocytes predominantly express Gαi2 and Gαi3, but not Gαi1. B or T lymphocytes lacking Gαi2 have poor chemotactic responses to chemokines, LN homing, adherence to high endothelial venules (HEV), and decreased motility within LN follicles, suggesting that Gαi3 does not compensate for the loss of Gαi2 in these processes (23,24). B lymphocytes lacking Gαi3 exhibit better chemotaxis than wild-type (WT) cells in response to gradients of multiple chemokines including CXCL12, CXCL13, and CCL19, and mice deficient in both Gαi2/3 have severely reduced numbers of B cells in the thymus, lymphoid organs, and gastrointestinal (GI) tract mucosa, and a hyper IgM-like syndrome with elevated levels of serum IgM but reduced levels of IgA, IgE, and IgG subclasses (25).
In studies of mice with the G184S mutation in Gαi2 (G184S KI), whose bone marrow was transplanted into WT donors, transplanted B cells had increased spontaneous migration and baseline intracellular Ca2+ levels in vitro but surprisingly poor chemotaxis and Ca2+ flux in response to several chemokines including CXCL12, CXCL13, and CCL19. B cell trafficking in vivo was significantly perturbed at baseline and following immunization, with increased B cell numbers in the spleen and reduced numbers in blood and other peripheral lymphoid tissues. Within the spleen, the mutation resulted in increased numbers of poorly formed germinal centers (GCs) (26).
Neutrophils from G184S KI mice also exhibited aberrant trafficking in vivo. In a sterile peritonitis model induced by intraperitoneal injection of thioglycollate, KI neutrophils showed reduced migration from the bone marrow to the bloodstream and from bloodstream to the peritoneum compared to WT controls. Increased bone marrow retention of mature neutrophils appeared to underlie these findings, presumably due to hyperactive basal activation (cytosolic Ca2+) and spontaneous migration but poor response to chemokine stimulation (27).
Collectively, these studies emphasize the importance of leukocyte-intrinsic chemokine GPCRs, Gαi proteins, and RGS protein activity for trafficking within the hematopoietic compartment under homeostatic conditions and for their capacity to migrate to tissues. In the remainder of this review, we summarize what is known about each individual R4 family member in the context of normal and pathological immune responses and/or inflammatory diseases, primarily based on studies of mice with loss or gain-of-function alleles and/or primary human cells.
PHYSIOLOGICAL FUNCTIONS OF INDIVIDUAL R4 RGS PROTEINS IN THE CELLS OF THE IMMUNE SYSTEM
RGS1—Controller of Lymphocyte and Monocyte/Macrophage Localization
Originally cloned as a B cell early activation gene (28), RGS1 is strongly expressed in immune cells including B lymphocytes (29,30), T lymphocytes (30,31), natural killer (NK) cells (31), dendritic cells (DCs) (32), and monocytes (33). Within the B cell compartment, RGS1 quantities are highest within GCs. These are micro-anatomic structures that develop within the secondary lymphoid organs in response to antigen stimulation, and formation of GCs is crucial for affinity maturation of antigen-specific B cells and their differentiation into antibody-secreting cells. Early studies showed that RGS1 regulates B cell homing to lymph nodes in mice and motility within the LN microenvironment by regulating chemokine signaling. Rgs1 −/− mice had spontaneous GC formation in spleens and more robust and prolonged responses to immunization (34). B lymphocytes from Rgs1 −/− mice transferred to WT recipients homed excessively to high endothelial venules (HEVs) of lymph nodes and transited through the lymph nodes at a faster rate than did WT cells (23), indicating that these abnormalities were due to intrinsic defects in B lymphocyte trafficking. Accordingly, RGS1-deficient B cells exhibited increased chemotaxis and accentuated and prolonged Ca2+ mobilization responses to chemokines including CXL12 (34).
More recently, the relationship between RGS1, its cognate partner Gαi2, and control of B lymphocyte trafficking was explored through additional studies of mice containing loss-of-function alleles of either or both genes. Loss of one or more alleles of Rgs1 enhanced chemotaxis of splenic B cells to multiple chemokines in vitro as expected. In contrast, loss of one or more Gnai2 alleles virtually disabled chemotactic responses of the B cells, which could be partially compensated for by loss of one or more alleles of Rgs1. Within the spleen, loss of RGS1 led to the presence of more follicles with spontaneously formed GCs containing increased numbers of marginal zone B cells; loss of Gnai2 alleles led to fewer follicles containing GCs and decreased marginal zone B cells, and double knockouts more closely resembled Rgs1 −/− mice. Thus, lymphoid organ follicle organization—crucial to the humoral immune response—is highly sensitive to the proportion of RGS1 and Gαi2 expression in peripheral lymphoid tissue (35).
Although splenic T cells from Rgs1 −/− mice exhibit chemotaxis equivalent to WT (34), studies have implicated a role for RGS1 and Gαi2 in T lymphocyte migration, particularly within the GI tract. Previous studies had demonstrated that Gnai2 −/− mice develop spontaneous inflammatory bowel disease resembling ulcerative colitis in humans (36). In both humans and mice, RGS1 is highly expressed in CD4+ and CD8+ T lymphocytes isolated from the GI mucosa relative to the analogous T cell subsets obtained from peripheral blood (37). Overexpression of RGS1 (but not RGS10) in peripheral blood T cells limited their chemotactic responses to CXCL12 and CCL19. Conversely, gut-derived T cells from Rgs1 −/− mice displayed increased chemotaxis to CXCL12 and CCL19 but not gut homing-associated chemokines CCL25 and CCL20 (ligands of CCR9 and CCR6, respectively). In a model of colitis induced by intraperitoneal injection of CD4+ T cells, Rag2 −/− (T and B cell-deficient) mice receiving T cells from Rgs1 −/− mice had ameliorated disease relative to mice receiving WT cells. Together, these findings indicate that the balance between expression of Gαi2 and RGS1 in gut T cells may control their retention within the GI mucosa.
RGS1 is also expressed in both monocytes and tissue macrophages, and recent studies of atherosclerosis-prone mice have implicated RGS1 in macrophage-mediated vascular inflammation associated with atherosclerotic plaques. Macrophages express receptors for proinflammatory chemokine receptors including CCR2 and CCR5, and expression is notably increased in aortic-associated macrophages from aged mice deficient in apolipoprotein E (ApoE −/−), a standard model for atherosclerosis, suggesting a pathogenic role for these chemokines (38). Proinflammatory signals (lipopolysaccharide [LPS] or IFNγ) upregulate RGS1 in mouse bone marrow-derived macrophages (39), and increased Rgs1 mRNA quantities were detected in macrophages isolated from both human carotid and abdominal aortic atherosclerotic plaques relative to those from non-diseased arteries or peripheral blood monocytes (40). While chemotaxis of macrophages from Rgs1 −/− /ApoE −/− mice to CCR2 and CCR5 ligands in vitro was significantly increased, the loss of RGS1 protected mice from the development of aortic atherosclerotic plaque formation in the aortic root of ApoE −/− mice. Underlying this protection appeared to be poor retention of RGS1-deficient macrophages to atherosclerotic plaques. Although early recruitment of these leukocytes to plaques (induced by continuous infusion of angiotensin II [AngII]) was similar in WT and Rgs1 −/−mice, the number of retained macrophages in plaques was reduced 10-fold in Rgs1 −/− mice compared to WT over time, and surface CCR2 expression was also increased. These findings suggest poor chemokine receptor desensitization due to prolonged signaling in the absence of RGS1, leading to transit of macrophages from plaques. Thus, similar to GI pathology in the absence of RGS1, the poor retention of a leukocyte population in the absence of a brake provided by RGS1 served an anti-inflammatory function in this setting.
Finally, a growing number of large genome-wide association studies (GWAS) in humans have identified a link between polymorphic variants in Rgs1 and chronic inflammatory diseases including celiac disease, multiple sclerosis, and type I diabetes (41–43). Multiple sclerosis (MS) is a relapsing-remitting degenerative disease of the central nervous system (CNS) white matter primarily driven by the influx of inflammatory T helper type 1 (TH1) and T helper type 17 (TH17) lymphocytes producing IFNγ and interleukin (IL)-17, respectively (44). GWAS have implicated single-nucleotide polymorphisms (SNPs) in Rgs1 in the pathogenesis of MS. SNPs rs1323292_A (odds ratio [OR] 1.12; p < 8.8 × 10−7) or rs2760524_A (OR 0.87, p < 3.6 × 10−9) were identified as susceptibility genes for MS (45–47). The latter allele also conferred susceptibility to celiac disease, a chronic GI inflammatory disease associated with sensitivity to gluten (42). Although RGS1 expression was similar in peripheral blood mononuclear cells (PBMCs) from patients with MS and controls (48), a separate study showed that modulation of RGS1 expression by interferon-β (IFN-β), a standard treatment for MS (49,50). RGS1 was also upregulated in disease-promoting TH17 cells isolated from the CNS of mice in a model of human MS, experimental autoimmune encephalomyelitis (EAE) (51). Although definitive mechanistic studies of RGS1 in MS have yet to be performed, these findings suggest that high expression of RGS1 could promote retention of proinflammatory T cells in the CNS.
RGS2—Unique Regulator of Gαq
RGS2 is widely expressed in the cardiovascular system including the brain, heart, vasculature, kidney, vascular smooth muscle, and endothelial cells (52–54). RGS2 possesses somewhat unique biochemical properties among R4 RGS family members. Its GAP activity is relatively selective for Gαq in vitro, and it appears to predominantly regulate Gq-mediated signaling pathways in vivo (55). RGS2 also interacts with Gαs and adenylyl cyclase (AC), suppressing Gαs signaling pathways independently of GAP activity (56). RGS2 can exist in several isoforms as a result of alternate translation initiation sites. A gain-of-function mutation in human RGS2 (G32D) was reported to increase expression of the largest RGS2 isoform, which displays stronger binding to AC and more robust inhibition of AC activity than WT RGS2. Platelets from carriers of this mutation showed decreased cyclic AMP formation upon stimulation with prostacyclin, suggesting that RGS2 is a negative regulator of Gs signaling in platelets (57). In contrast, Rgs2 −/− mice had no defects in megakaryopoiesis and vascular injury-induced thrombus formation equivalent to control mice, suggesting that RGS2 is not a predominant regulator of hemostasis in mice (58).
RGS2 has functions in both T lymphocyte proliferation and cytokine production. T cells from the lymph nodes of naïve Rgs2 −/− mice had markedly reduced proliferation and IL-2 production after phorbol ester/Ca2+ ionophore or T cell receptor (anti-CD3/CD28) stimulation in vitro compared to WT whereas B cell proliferation induced by antigen receptor stimulation (anti-IgM) was intact, and serum immunoglobulin levels were normal (59). Although inflammation (footpad swelling) induced by injection of lymphocytic choriomeningitis virus (LCMV) was significantly reduced in Rgs2 +/− mice compared to knockouts, detailed analysis of the composition of inflammatory infiltrates was not performed in this study. In addition, migration and/or signaling (presumably Ca2+mobilization) of both CD4+ and CD8+ T cells from Rgs2 +/− mice and Rgs2 +/− mice were reported to be equivalent in response to multiple ligands (e.g., chemokines, histamine, lysophosphatidic acid). Thus, the precise role of RGS2 in T lymphocyte physiology remains to be determined.
RGS13—Potential Roles in Autoimmunity, Lymphoid Malignancy, and Allergic Responses
RGS13 is one of the smallest RGS proteins in R4 subfamily, containing only a short alpha helix domain at the N-terminus in addition to the shared RGS box. Human and mouse RGS13, cloned and characterized in 2002, (60,61) are composed of 159 and 157 amino acids, respectively, with 82% identity. RGS13 is expressed mainly in immune cells including GC B and T lymphocytes and mast cells (61–64). In human diseases, RGS13 is also highly expressed in Burkitt’s lymphoma, a malignancy of GC B cells, and adult T cell leukemia/lymphoma (ALT), but nearly absent in mantle cell lymphoma (65–67). RGS13 was also recently linked to asthma by virtue of a higher expression in peripheral blood T regulatory cells from children with non-allergic asthma compared to controls (68).
As noted above, follicular B cells follow a CXCL12/13 gradient to migrate from the follicle edge to the follicle center to form a nascent GC. The mature GC consists of two distinct anatomical regions—the dark zone, where B cells first encounter antigen and undergo proliferation, and somatic hypermutation of their immunoglobulin (Ig) genes to generate high-affinity antigen receptors and the light zone, where they undergo antibody class switching and affinity maturation through interactions with follicular helper T (TFH) cells and follicular dendritic cells (FDC). CXCL12 and CXCL13 and their cognate receptors CXCR4 and CXCR5 drive B cell entrance and transit within GCs, and perturbed B cell-T cell or B cell-FDC interactions may result in autoimmunity. Rgs13 −/− mice had enlarged GCs in spleen and more robust antibody responses than WT mice following immunization (69). In BXD2 mice, which are a model of systemic autoimmunity associated with spontaneous GC formation and pathogenic autoantibodies, RGS13 and RGS16 are upregulated in GC B cells and TFH cells, respectively, in an IL-17-dependent manner (70). GCs from BXD2 mice have increased numbers of plasmablasts, expression of B cell differentiation genes, and TFH populations. B cell and T cell abnormalities in this strain are rescued by gene deletion of Rgs13 or Rgs16, respectively, which is accompanied by significantly reduced numbers of B-T cell conjugates in GCs and autoantibody titers (71,72). Given RGS13’s ability to blunt chemokine-mediated migration, these studies suggest that IL-17 promotes autoimmunity in BXD2 mice by prolonging transit of B and T cells through GCs, resulting in excess antibody production.
A second, non-canonical function of RGS13 in the regulation of gene expression may also affect B cell functions in lymphoid organs. RGS13 translocates to the nucleus upon stimulation of adrenergic receptors, where it binds phosphorylated cyclic AMP response element binding protein (CREB), thereby inhibiting CREB-mediated transcription by disrupting promoter DNA-protein complexes (73). Several CREB target genes important for GC B cell physiological functions, including c-fosb and Obf1, were upregulated in GC B cells from Rgs13 −/− mice (72).
Finally, RGS13 is also abundantly expressed in human and mouse mast cells (MCs). MCs—tissue-based innate immune cells located in close proximity to the nerves and blood vessels—degranulate upon crosslinking of high-affinity IgE receptors (FcεRI) by allergens (which are defined as innocuous substances in non-allergic people). Classical allergic responses can be initiated following MC degranulation through release of preformed and newly synthesized mediators of anaphylaxis (e.g., histamine, leukotrienes). Knockdown of RGS13 in human MC lines enhanced GPCR-mediated Ca2+ mobilization and chemotaxis presumably through GAP-dependent mechanisms (64). Surprisingly, however, RGS13 was also found to inhibit IgE-mediated MC degranulation, which was mediated by a direct interaction with a key signaling molecule downstream of FcεRI—phosphatidylinositol-3-OH kinase (PI3 kinase). RGS13 (and several other members of the R4 RGS subfamily) physically interacted with the regulatory p85α subunit of PI3K and limited its association with a membrane signalosome. As a result, Rgs13 −/− mice had increased IgE-induced systemic anaphylaxis. In sum, these specific functions of RGS13 and its highly restricted expression in GC B cells and MCs make it an attractive potential drug target for the treatment of autoantibody-associated and/or allergic diseases.
RGS16—Guidepost for Inflammatory T Cell and Monocyte Functions
RGS16 was initially cloned from the retina (74), and subsequent studies demonstrated widespread tissue expression. In blood cells, RGS16 has been found in NK cells (31), platelets (75), dendritic cells (32), and T lymphocytes (76). Several studies suggest a role for RGS16 in allergic airway inflammation that frequently accompanies asthma in humans. Inhalation of environmental allergens leads to production of allergen-specific IgE and expansion and activation of TH2 and type 2 innate lymphoid cells; production of allergic cytokines IL-4, 5, 13, 25, and 33; eosinophil recruitment to the airways; and increased mucus secretion by goblet cells (77). Cumulatively, these proinflammatory changes result in hyperresponsiveness of the airways to contraction triggers (“airway hyperresponsiveness” [AHR]). In early studies of T cell-specific Rgs16 transgenic mice, T cell trafficking to the lung was reduced in a model of allergic airway inflammation, but there were substantially more cytokine-producing TH2 cells in the lymphoid organs, resulting in increased systemic inflammation and AHR (78). The lungs of Rgs16 −/− mice challenged with allergen contained significantly more TH2 cells than those of WT mice following allergen challenge, suggesting that RGS16 regulates lung inflammation by directing both T lymphocyte trafficking into tissues and cytokine production (79).
RGS16 overexpression in human monocyte THP-1 cells stimulated with LPS decreased production of proinflammatory cytokines IL-1β, IL-6, and TNFα, while RGS16 knockdown by RNAi had the opposite effect (80). Rgs16 transcripts were upregulated more than 100-fold in human monocyte-derived DCs treated with LPS and IL-10 compared to untreated cells (81). The ORF3 protein from porcine circovirus type 2 (PCV2), an important cause of systemic wasting in pigs, was found to bind specifically to porcine RGS16 (closely related to human and murine RGS16), and expression of porcine RGS16 was induced by several cellular activators including LPS (82), suggesting a possible role of RGS16 in PCV2 infection. More recent work demonstrated that ORF3 binding to RGS16 in transfected cells increases RGS16 degradation and enhances NFκB activation and transcription of proinflammatory cytokine genes (Il6 and Il8) (83). This is a prime example of pathogens subverting the host immune response and promoting inflammation by regulating G protein-mediated signaling.
RGS18—Controller of Platelet Functions
RGS18 expression appears to be relatively restricted to bone marrow-derived cells (84–86). RGS18 is abundantly and predominantly expressed in platelets and, to a lesser extent in megakaryocytes (MKs) and leukocytes, but not in erythrocytes (87). Promoter analysis at the Rgs18 locus demonstrated highly restricted occupancy of both GATA-1 and GATA-2 transcription factors, which may partially explain its enrichment in the hematopoietic compartment (88). Overexpression experiments have confirmed that RGS18 is capable of inhibiting both Gi- and Gq-mediated signaling pathways. However, RGS18 overexpression had no effect on CXCR4 signaling in one study (89). RGS18 is also expressed in osteoclasts (90)—bone-resorbing multinuclear giant cells derived from macrophage/monocyte lineage precursors, which play crucial roles in bone remodeling. Osteoclast differentiation is controlled by RANK ligand (receptor activator of nuclear factor κB ligand), which acts on the RANK receptor to induce osteoclast differentiation. Upregulation of a GPCR that senses extracellular acidosis, OGR1, leads to proton stimulation of OGR1 and activation the Gq-PLCβ pathway, which in turn increases nuclear factor of activated T cells (NFAT) activity and promotes osteoclastogenesis. RANKL reduced RGS18 expression in the osteoclast precursor cell line RAW246.7 and in primary bone marrow-derived osteoclast precursor monocytes. RGS18 knockdown in RAW246.7 cells (by RNAi) enhanced osteoclast differentiation evoked by RANKL, and an anti-OGR1 blocking antibody neutralized this phenotype. Further, RGS18 knockdown increased NFAT activation induced by extracellular acidosis, while RGS18 (but not RGS2 or GAIP) overexpression inhibited OGR1-mediated responses. Taken together, the studies indicate that RGS18 levels may control osteoclastogenesis mediated by RANKL through modulation of OGR1-evoked signaling.
Recent studies have focused on the role of RGS18 in megakaryopoiesis and/or platelet function. Gegenbauer et al. showed that RGS18 is a target of PKA- and PKG-mediated platelet inhibition and GPCR-mediated platelet activation. RGS18 is phosphorylated on serine 49 and serine 218 upon platelet activation with activators such as thrombin or thromboxane A2, which promotes binding to 14-3-3γ, a phosphoserine-binding scaffolding protein expressed in platelets, which inhibits RGS18 GAP activity. In contrast, physiological platelet inhibitors PGI2 (through cyclic AMP and protein kinase A) and nitric oxide (NO) (through cyclic GMP and protein kinase G) cause phosphorylation of Ser216, which displaces 14-3-3γ from RGS18 and enhances its inhibition of G protein-dependent signaling (91). Rgs18 −/− mice are mildly thrombocytopenic due to decreased megakaryopoiesis but are also hypercoagulable due to increased platelet responsiveness in vitro, resulting in increased thrombus formation in vivo (92,93). Thus, these early studies suggest that RGS18 is a critical modulator of both megakaryocyte differentiation and hemostatic functions of mature platelets.
RGS3, 4, 5, 8, 21—Few Known Functions in the Immune System
Little is known about the specific physiological functions of these particular RGS proteins in the immune system, either because of insufficient study or tissue expression primarily confined to non-immune cells. As we have seen with RGS1 and 2, however, RGS protein expression can be transiently upregulated (or downregulated) in immune cells in specific settings—an inflammatory microenvironment being the prime example. A study of rat leukocytes revealed constitutive expression of Rgs5 mRNA in CD4+ T lymphocytes and Rgs8 in natural killer (NK) cells (31), and Rgs2, Rgs16, and Rgs18 expression was induced by crosslinking NK inhibitory Ly49a or Ly49D receptors, which recognize MHC Class I. This is yet another example of the need for detailed examination of RGS expression in the appropriate context to determine their potential contribution to physiological immune responses or inflammatory pathology.
RGS3 is unique among R4 subfamily members, in that, in addition to having the requisite RGS box, its PDZ domain mediates binding to a distinct set of membrane-associated signaling proteins. PDZ-RGS3 interacts with B class ephrins, which mediate fundamental processes in developing neurons including migration and axon guidance through interaction with ephrin receptor tyrosine kinases on neighboring cells (94,95). RGS3 is expressed widely, and perhaps as a result, global knockout of PDZ-RGS3 leads to increased rates of embryonic lethality associated with abnormalities in the CNS development (95).
Like RGS16, a potential role for RGS3 in the recruitment of inflammatory T cells was demonstrated in a mouse model of asthma. RGS3 was expressed in both TH1 and TH2 cells differentiated from naïve splenic T lymphocytes in vitro, and overexpression or knockdown of RGS3 modulated chemotaxis of a T cell line to CXCL12 (96). In mice expressing alleles containing a targeted deletion of the RGS domain of RGS3 (Rgs3 ΔRGS), architecture of the thymus and peripheral lymphoid organs and B and T lymphocyte numbers in the periphery were relatively similar to WT mice at baseline. However, following sensitization and respiratory inoculation with house dust mite allergen, Rgs3 ΔRGS mice had exaggerated allergen-induced lung inflammation, with increased numbers of activated T cells and formation of perivascular structures resembling bronchus-associated lymphoid tissue (BALT) (96). Thus, like RGS16, RGS3 may regulate proinflammatory T cell trafficking in response to immune challenge.
RGS4 is expressed constitutively in the highest amounts in the brain and heart in both humans and mice; genetic deletion of Rgs4 in mice results in cardiovascular abnormalities including conduction defects in the heart and a propensity for development of arrhythmias (97). Rgs4 −/− mice also have altered nociceptive (pain) responses and tolerance to opioid analgesics and antidepressants (98,99). However, detailed analysis of RGS4 expression and function in leukocyte subsets has not been performed nor have prominent immune system abnormalities been reported in mice deficient in RGS4.
RGS5 is abundantly expressed in the heart and Rgs5 −/− mice exhibit cardiovascular abnormalities and arrhythmias (100–102). RGS5 is also expressed in macrophages and, like RGS1, may regulate atherosclerotic plaque formation in AngII-induced and ApoE −/− mouse models of atherosclerosis. However, in contrast to RGS1, the loss of RGS5 exacerbated the development of atherosclerosis ApoE −/− mice due to increased inflammatory mediator (IL-1β, TNFα, IL-6) expression in diseased vessels and increased accumulation of apoptotic macrophages, endothelial cells, and lipids in the plaque wall (103). A separate study showed that RGS5 is expressed in peritoneal macrophages and is downregulated by AngII. Agonists of the anti-inflammatory peroxisome proliferator-activated receptor (PPAR) family of nuclear receptors counteracted proinflammatory signaling evoked by AngII (Erk phosphorylation, c-fos expression), which was reversed by RGS5 knockdown (104), suggesting that PPAR agonists exert anti-atherogenic effects on macrophages by regulating RGS5 expression. It will be of interest to determine the relative role(s) of RGS1 and RGS5 in proinflammatory macrophages and vascular cells (i.e., endothelial cells, pericytes) through definitive studies of mice with genetic deletion of one or more alleles in models of atherosclerosis and other inflammatory vascular disorders.
RGS8 is abundantly expressed in brain, with particular enrichment in cerebellar Purkinje cells, but its physiological functions have not been well characterized (105). Early studies showed that RGS8 actually enhances kinetics of signaling by G protein-gated potassium channels in the presence of an agonist, perhaps by acting as a scaffold that facilitates G protein nucleotide cycling (106,107). A recent RNAi screen demonstrated that knockdown of RGS8 uniquely reduced agonist-induced Ca2+ mobilization induced by activation of thrombin and m3 muscarinic receptors, in contrast to siRNAs targeting all other R4 RGSs, which may be due to reduced GPCR expression on the membrane (108). Thus, RGS8 may have pleiotropic functions in GPCR-mediated signaling not shared by other R4 family members. Although Rgs8 transcripts were detected in rat NK cells (31), its functions in the immune system have not been explored.
RGS21, which is the most recently discovered and the smallest member of the R4 RGS family (109,110), is abundantly expressed in the olfactory and lingual epithelia and in the sinoatrial mucosal tissue (111). RGS21 appears to have GAP activity similar to the other R4 isoforms. Although Rgs21 transcripts were readily detected in multiple tissues tested including the spleen, thymus, peripheral blood leukocytes, and GI tract in one study, its physiological role in the immune system has not yet been investigated (110).
Conclusions: Can R4 RGS Proteins Be Targeted for the Treatment of Inflammatory Diseases?
The published work discussed here suggests prominent roles for R4 RGS proteins in lymphocyte and macrophage trafficking to and from sites of tissue inflammation and B cell maturation occurring in transit through lymphoid organs, which is required for effective antibody-mediated adaptive immune responses. Although functions of R4 RGS proteins in innate immune cells—including granulocytes (neutrophils, eosinophils, basophils), NK, NKT, and innate lymphoid cells—are virtually unknown, it is likely that they regulate migration and localization of these leukocytes to varying degrees.
Whether inhibiting or boosting RGS function would be desirable for therapy of a particular inflammatory or autoimmune disease is uncertain, but recent studies support the feasibility of either approach. Thiadiazolidinone (TDZD) inhibitors of RGS4 with nanomolar potency and selectivity (8- to >5000-fold) for RGS4 over other RGS proteins have been pioneered by Neubig and colleagues (112). Small molecule compounds such as CCG50014 inhibit RGS4-G protein interactions by covalently modifying cysteine residues in the RGS box similar to physiological by-products of oxidative stress metabolism (4-hydroxy-2-nonenal [4HNE]) (113). CCG50014 has been used in rodents in vivo in a recent study; intrathecal administration to mice augmented opioid analgesic responses in a dose-dependent manner (114).
Pharmacological enhancers of RGS activity have not yet been discovered, but tactics designed to selectively increase RGS expression might represent a viable option. Several R4 RGS proteins including RGS4 and 5 are substrates of the arginine (Arg)/N-end rule pathway, which targets proteins for proteasomal degradation through acetylation of positively charged N-terminal residues (115). Administration of the neurostimulant, para-chloroamphetamine (PCA), an inhibitor of the Arg/N-end rule pathway, to mice intrathecally increased RGS4 expression in the brain 300-fold and blunted activation of GPCR signaling (116). In a separate study, proteasomal inhibitors such as bortezomib also increased RGS4 expression (115). Topical or mucosal (e.g., inhalational) administration of such compounds could increase their efficacy in inflammatory diseases of the mucosa (e.g., asthma), while limiting systemic side effects. Finally, a recent cell-based HTS of a microbial-derived natural compound library identified indolactam V as a specific upregulator of RGS2 but not RGS4, through a protein kinase C (PKC)-mediated mechanism, supporting the feasibility of this experimental strategy (117).
These examples illustrate the potential of suppression or augmentation of RGS levels and/or activity to improve efficacy of GPCR-targeted drugs, which could ultimately limit side effects and prevent tolerance. Collectively, these studies provide a proof-of-concept foundation for the development of selective modulators of individual RGS proteins for the treatment of an array of diseases including many of the inflammatory diseases discussed here.
DC, dendritic cell; FDC, follicular dendritic cell; GC, germinal center; GI, gastrointestinal; GPCR, G protein-coupled receptor; MC, mast cell; NK, natural killer; RGS, the regulator of signaling; TFH, follicular helper T.
References
Sellge G, Kufer TA. PRR-signaling pathways: learning from microbial tactics. Semin Immunol. 2015;27:75–84.
Zugasti O, Bose N, Squiban B, Belougne J, Kurz CL, Schroeder FC, et al. Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of Caenorhabditis elegans. Nat Immunol. 2014;15:833–8.
Bloes DA, Kretschmer D, Peschel A. Enemy attraction: bacterial agonists for leukocyte chemotaxis receptors. Nat Rev Microbiol. 2015;13:95–104.
Lian J, Luster AD. Chemokine-guided cell positioning in the lymph node orchestrates the generation of adaptive immune responses. Curr Opin Cell Biol. 2015;36:1–6.
Anders HJ, Romagnani P, Mantovani A. Pathomechanisms: homeostatic chemokines in health, tissue regeneration, and progressive diseases. Trends Mol Med. 2014;20:154–65.
Arnon TI, Cyster JG. Blood, sphingosine-1-phosphate and lymphocyte migration dynamics in the spleen. Curr Top Microbiol Immunol. 2014;378:107–28.
Gilman AG. G proteins: transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–49.
Beck TC, Gomes AC, Cyster JG, Pereira JP. CXCR4 and a cell-extrinsic mechanism control immature B lymphocyte egress from bone marrow. J Exp Med. 2014;211:2567–81.
Mocsai A, Walzog B, Lowell CA. Intracellular signalling during neutrophil recruitment. Cardiovasc Res. 2015;107:373–85.
Packiriswamy N, Parameswaran N. G-protein-coupled receptor kinases in inflammation and disease. Genes Immun. 2015;16(6):367–77.
Clister T, Mehta S, Zhang J. Single-cell analysis of G-protein signal transduction. J Biol Chem. 2015;290:6681–8.
Kimple AJ, Bosch DE, Giguere PM, Siderovski DP. Regulators of G-protein signaling and their Galpha substrates: promises and challenges in their use as drug discovery targets. Pharmacol Rev. 2011;63:728–49.
Sjogren B, Blazer LL, Neubig RR. Regulators of G protein signaling proteins as targets for drug discovery. Prog Mol Biol Transl Sci. 2010;91:81–119.
Bansal G, Druey KM, Xie Z. R4 RGS proteins: regulation of G-protein signaling and beyond. Pharmacol Ther. 2007;116:473–95.
Heximer SP, Srinivasa SP, Bernstein LS, Bernard JL, Linder ME, Hepler JR, et al. G protein selectivity is a determinant of RGS2 function. J Biol Chem. 1999;274:34253–9.
Kach J, Sethakorn N, Dulin NO. A finer tuning of G-protein signaling through regulated control of RGS proteins. Am J Physiol Heart Circ Physiol. 2012;303:H19–35.
Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein alpha subunits. Cell. 1996;86:445–52.
Kimple AJ, Willard FS, Giguere PM, Johnston CA, Mocanu V, Siderovski DP. The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4 Galpha-interaction face. Biochim Biophys Acta. 2007;1774(9):1213–20.
DiBello PR, Garrison TR, Apanovitch DM, Hoffman G, Shuey DJ, Mason K, et al. Selective uncoupling of RGS action by a single point mutation in the G protein alpha-subunit. J Biol Chem. 1998;273:5780–4.
Huang X, Charbeneau RA, Fu Y, Kaur K, Gerin I, MacDougald OA, et al. Resistance to diet-induced obesity and improved insulin sensitivity in mice with a regulator of G protein signaling-insensitive G184S Gnai2 allele. Diabetes. 2008;57:77–85.
Kaur K, Kehrl JM, Charbeneau RA, Neubig RR. RGS-insensitive Galpha subunits: probes of Galpha subtype-selective signaling and physiological functions of RGS proteins. Methods Mol Biol. 2011;756:75–98.
Neubig RR. RGS-insensitive G proteins as in vivo probes of RGS function. Prog Mol Biol Transl Sci. 2015;133:13–30.
Han SB, Moratz C, Huang NN, Kelsall B, Cho H, Shi CS, et al. Rgs1 and Gnai2 regulate the entrance of B lymphocytes into lymph nodes and B cell motility within lymph node follicles. Immunity. 2005;22:343–54.
Hwang IY, Park C, Kehrl JH. Impaired trafficking of Gnai2+/− and Gnai2−/− T lymphocytes: implications for T cell movement within lymph nodes. J Immunol. 2007;179:439–48.
Hwang IY, Park C, Luong T, Harrison KA, Birnbaumer L, Kehrl JH. The loss of Gnai2 and Gnai3 in B cells eliminates B lymphocyte compartments and leads to a hyper-IgM like syndrome. PLoS One. 2013;8, e72596.
Hwang IY, Park C, Harrison K, Boularan C, Gales C, Kehrl JH. An essential role for RGS protein/Galphai2 interactions in B lymphocyte-directed cell migration and trafficking. J Immunol. 2015;194:2128–39.
Cho H, Kamenyeva O, Yung S, Gao JL, Hwang IY, Park C, et al. The loss of RGS protein-Galpha(i2) interactions results in markedly impaired mouse neutrophil trafficking to inflammatory sites. Mol Cell Biol. 2012;32:4561–71.
Hong JX, Wilson GL, Fox CH, Kehrl JH. Isolation and characterization of a novel B cell activation gene. J Immunol. 1993;150:3895–904.
Moratz C, Kang VH, Druey KM, Shi CS, Scheschonka A, Murphy PM, et al. Regulator of G protein signaling 1 (RGS1) markedly impairs Gi alpha signaling responses of B lymphocytes. J Immunol. 2000;164:1829–38.
Agenes F, Bosco N, Mascarell L, Fritah S, Ceredig R. Differential expression of regulator of G-protein signalling transcripts and in vivo migration of CD4+ naive and regulatory T cells. Immunology. 2005;115:179–88.
Kveberg L, Ryan JC, Rolstad B, Inngjerdingen M. Expression of regulator of G protein signalling proteins in natural killer cells, and their modulation by Ly49A and Ly49D. Immunology. 2005;115:358–65.
Shi GX, Harrison K, Han SB, Moratz C, Kehrl JH. Toll-like receptor signaling alters the expression of regulator of G protein signaling proteins in dendritic cells: implications for G protein-coupled receptor signaling. J Immunol. 2004;172:5175–84.
Denecke B, Meyerdierks A, Bottger EC. RGS1 is expressed in monocytes and acts as a GTPase-activating protein for G-protein-coupled chemoattractant receptors. J Biol Chem. 1999;274:26860–8.
Moratz C, Hayman JR, Gu H, Kehrl JH. Abnormal B-cell responses to chemokines, disturbed plasma cell localization, and distorted immune tissue architecture in Rgs1−/− mice. Mol Cell Biol. 2004;24:5767–75.
Hwang IY, Park C, Harrision KA, Huang NN, Kehrl JH. Variations in Gnai2 and Rgs1 expression affect chemokine receptor signaling and the organization of secondary lymphoid organs. Genes Immun. 2010;11:384–96.
Rudolph U, Finegold MJ, Rich SS, Harriman GR, Srinivasan Y, Brabet P, et al. Ulcerative colitis and adenocarcinoma of the colon in G alpha i2-deficient mice. Nat Genet. 1995;10:143–50.
Gibbons DL, Abeler-Dorner L, Raine T, Hwang IY, Jandke A, Wencker M, et al. Cutting edge: regulator of G protein signaling-1 selectively regulates gut T cell trafficking and colitic potential. J Immunol. 2011;187:2067–71.
Wan W, Murphy PM. Regulation of atherogenesis by chemokines and chemokine receptors. Arch Immunol Ther Exp (Warsz). 2013;61:1–14.
Riekenberg S, Farhat K, Debarry J, Heine H, Jung G, Wiesmuller KH, et al. Regulators of G-protein signalling are modulated by bacterial lipopeptides and lipopolysaccharide. FEBS J. 2009;276:649–59.
Patel J, McNeill E, Douglas G, Hale AB, de Bono J, Lee R, et al. RGS1 regulates myeloid cell accumulation in atherosclerosis and aortic aneurysm rupture through altered chemokine signalling. Nat Commun. 2015;6:6614.
Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–77.
Hunt KA, Zhernakova A, Turner G, Heap GA, Franke L, Bruinenberg M, et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat Genet. 2008;40:395–402.
Johnson BA, Wang J, Taylor EM, Caillier SJ, Herbert J, Khan OA, et al. Multiple sclerosis susceptibility alleles in African Americans. Genes Immun. 2010;11:343–50.
Comabella M, Khoury SJ. Immunopathogenesis of multiple sclerosis. Clin Immunol. 2012;142:2–8.
International Multiple Sclerosis Genetics, C. IL12A, MPHOSPH9/CDK2AP1 and RGS1 are novel multiple sclerosis susceptibility loci. Genes Immun. 2010;11:397–405.
International Multiple Sclerosis Genetics, C, Wellcome Trust Case Control, C, Sawcer S, Hellenthal G, Pirinen M, Spencer CC, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–9.
Habek M, Brinar VV, Borovecki F. Genes associated with multiple sclerosis: 15 and counting. Expert Rev Mol Diagn. 2010;10:857–61.
Perga S, Montarolo F, Martire S, Berchialla P, Malucchi S, Bertolotto A. Anti-inflammatory genes associated with multiple sclerosis: a gene expression study. J Neuroimmunol. 2015;279:75–8.
Wostradowski T, Gudi V, Pul R, Gingele S, Lindquist JA, Stangel M, et al. Effect of IFN beta-1b on CXCR4-dependent chemotaxis in T cells from multiple sclerosis patients. Clin Exp Immunol. 2015;182(2):162–72.
Tran T, Paz P, Velichko S, Cifrese J, Belur P, Yamaguchi KD, et al. Interferonbeta-1b induces the expression of RGS1 a negative regulator of G-protein signaling. Int J Cell Biol. 2010;2010:529376.
Hoppmann N, Graetz C, Paterka M, Poisa-Beiro L, Larochelle C, Hasan M, et al. New candidates for CD4 T cell pathogenicity in experimental neuroinflammation and multiple sclerosis. Brain. 2015;138:902–17.
Heximer SP, Knutsen RH, Sun X, Kaltenbronn KM, Rhee MH, Peng N, et al. Hypertension and prolonged vasoconstrictor signaling in RGS2-deficient mice. J Clin Invest. 2003;111:445–52.
Wang X, Adams LD, Pabon LM, Mahoney Jr WM, Beaudry D, Gunaje J, et al. RGS5, RGS4, and RGS2 expression and aortic contractibility are dynamically co-regulated during aortic banding-induced hypertrophy. J Mol Cell Cardiol. 2008;44:539–50.
Kehrl JH, Sinnarajah S. RGS2: a multifunctional regulator of G-protein signaling. Int J Biochem Cell Biol. 2002;34:432–8.
Nance MR, Kreutz B, Tesmer VM, Sterne-Marr R, Kozasa T, Tesmer JJ. Structural and functional analysis of the regulator of G protein signaling 2-galphaq complex. Structure. 2013;21:438–48.
Roy AA, Nunn C, Ming H, Zou MX, Penninger J, Kirshenbaum LA, et al. Up-regulation of endogenous RGS2 mediates cross-desensitization between Gs and Gq signaling in osteoblasts. J Biol Chem. 2006;281:32684–93.
Noe L, Di Michele M, Giets E, Thys C, Wittevrongel C, De Vos R, et al. Platelet Gs hypofunction and abnormal morphology resulting from a heterozygous RGS2 mutation. J Thromb Haemost. 2010;8:1594–603.
Banno F, Nojiri T, Matsumoto S, Kamide K, Miyata T. RGS2 deficiency in mice does not affect platelet thrombus formation at sites of vascular injury. J Thromb Haemost. 2012;10:309–11.
Oliveira-Dos-Santos AJ, Matsumoto G, Snow BE, Bai D, Houston FP, Whishaw IQ, et al. Regulation of T cell activation, anxiety, and male aggression by RGS2. Proc Natl Acad Sci U S A. 2000;97:12272–7.
Johnson EN, Druey KM. Functional characterization of the G protein regulator RGS13. J Biol Chem. 2002;277:16768–74.
Shi GX, Harrison K, Wilson GL, Moratz C, Kehrl JH. RGS13 regulates germinal center B lymphocytes responsiveness to CXC chemokine ligand (CXCL)12 and CXCL13. J Immunol. 2002;169:2507–15.
Estes JD, Thacker TC, Hampton DL, Kell SA, Keele BF, Palenske EA, et al. Follicular dendritic cell regulation of CXCR4-mediated germinal center CD4 T cell migration. J Immunol. 2004;173:6169–78.
Bansal G, Xie Z, Rao S, Nocka KH, Druey KM. Suppression of immunoglobulin E-mediated allergic responses by regulator of G protein signaling 13. Nat Immunol. 2008;9:73–80.
Bansal G, DiVietro JA, Kuehn HS, Rao S, Nocka KH, Gilfillan AM, et al. RGS13 controls g protein-coupled receptor-evoked responses of human mast cells. J Immunol. 2008;181:7882–90.
Islam TC, Asplund AC, Lindvall JM, Nygren L, Liden J, Kimby E, et al. High level of cannabinoid receptor 1, absence of regulator of G protein signalling 13 and differential expression of cyclin D1 in mantle cell lymphoma. Leukemia. 2003;17:1880–90.
Pise-Masison CA, Radonovich M, Dohoney K, Morris JC, O’Mahony D, Lee MJ, et al. Gene expression profiling of ATL patients: compilation of disease-related genes and evidence for TCF4 involvement in BIRC5 gene expression and cell viability. Blood. 2009;113:4016–26.
Sethakorn N, Dulin NO. RGS expression in cancer: oncomining the cancer microarray data. J Recept Signal Transduct Res. 2013;33:166–71.
Raedler D, Ballenberger N, Klucker E, Bock A, Otto R, Prazeres da Costa O, et al. Identification of novel immune phenotypes for allergic and nonallergic childhood asthma. J Allergy Clin Immunol. 2015;135:81–91.
Hwang IY, Hwang KS, Park C, Harrison KA, Kehrl JH. Rgs13 constrains early B cell responses and limits germinal center sizes. PLoS One. 2013;8, e60139.
Hsu HC, Yang P, Wang J, Wu Q, Myers R, Chen J, et al. Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol. 2008;9:166–75.
Ding Y, Li J, Wu Q, Yang P, Luo B, Xie S, et al. IL-17RA is essential for optimal localization of follicular Th cells in the germinal center light zone to promote autoantibody-producing B cells. J Immunol. 2013;191:1614–24.
Wang JH, New JS, Xie S, Yang P, Wu Q, Li J, et al. Extension of the germinal center stage of B cell development promotes autoantibodies in BXD2 mice. Arthritis Rheum. 2013;65:2703–12.
Xie Z, Geiger TR, Johnson EN, Nyborg JK, Druey KM. RGS13 acts as a nuclear repressor of CREB. Mol Cell. 2008;31:660–70.
Chen C, Zheng B, Han J, Lin SC. Characterization of a novel mammalian RGS protein that binds to Galpha proteins and inhibits pheromone signaling in yeast. J Biol Chem. 1997;272:8679–85.
Kim SD, Sung HJ, Park SK, Kim TW, Park SC, Kim SK, et al. The expression patterns of RGS transcripts in platelets. Platelets. 2006;17:493–7.
Beadling C, Druey KM, Richter G, Kehrl JH, Smith KA. Regulators of G protein signaling exhibit distinct patterns of gene expression and target G protein specificity in human lymphocytes. J Immunol. 1999;162:2677–82.
Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2015;16:45–56.
Lippert E, Yowe DL, Gonzalo JA, Justice JP, Webster JM, Fedyk ER, et al. Role of regulator of G protein signaling 16 in inflammation-induced T lymphocyte migration and activation. J Immunol. 2003;171:1542–55.
Shankar SP, Wilson MS, DiVietro JA, Mentink-Kane MM, Xie Z, Wynn TA, et al. RGS16 attenuates pulmonary Th2/Th17 inflammatory responses. J Immunol. 2012;188:6347–56.
Suurvali J, Pahtma M, Saar R, Paalme V, Nutt A, Tiivel T, et al. RGS16 restricts the pro-inflammatory response of monocytes. Scand J Immunol. 2015;81:23–30.
Perrier P, Martinez FO, Locati M, Bianchi G, Nebuloni M, Vago G, et al. Distinct transcriptional programs activated by interleukin-10 with or without lipopolysaccharide in dendritic cells: induction of the B cell-activating chemokine, CXC chemokine ligand 13. J Immunol. 2004;172:7031–42.
Timmusk S, Merlot E, Lovgren T, Jarvekulg L, Berg M, Fossum C. Regulator of G protein signalling 16 is a target for a porcine circovirus type 2 protein. J Gen Virol. 2009;90:2425–36.
Choi CY, Rho SB, Kim HS, Han J, Bae J, Lee SJ, et al. The ORF3 protein of porcine circovirus type 2 promotes secretion of IL-6 and IL-8 in porcine epithelial cells by facilitating proteasomal degradation of regulator of G protein signalling 16 through physical interaction. J Gen Virol. 2015;96:1098–108.
Nagata Y, Oda M, Nakata H, Shozaki Y, Kozasa T, Todokoro K. A novel regulator of G-protein signaling bearing GAP activity for Galphai and Galphaq in megakaryocytes. Blood. 2001;97:3051–60.
Park IK, Klug CA, Li K, Jerabek L, Li L, Nanamori M, et al. Molecular cloning and characterization of a novel regulator of G-protein signaling from mouse hematopoietic stem cells. J Biol Chem. 2001;276:915–23.
Yowe D, Weich N, Prabhudas M, Poisson L, Errada P, Kapeller R, et al. RGS18 is a myeloerythroid lineage-specific regulator of G-protein-signalling molecule highly expressed in megakaryocytes. Biochem J. 2001;359:109–18.
Gagnon AW, Murray DL, Leadley RJ. Cloning and characterization of a novel regulator of G protein signalling in human platelets. Cell Signal. 2002;14:595–606.
Johnson KD, Boyer ME, Kang JA, Wickrema A, Cantor AB, Bresnick EH. Friend of GATA-1-independent transcriptional repression: a novel mode of GATA-1 function. Blood. 2007;109:5230–3.
Berthebaud M, Riviere C, Jarrier P, Foudi A, Zhang Y, Compagno D, et al. RGS16 is a negative regulator of SDF-1-CXCR4 signaling in megakaryocytes. Blood. 2005;106:2962–8.
Iwai K, Koike M, Ohshima S, Miyatake K, Uchiyama Y, Saeki Y, et al. RGS18 acts as a negative regulator of osteoclastogenesis by modulating the acid-sensing OGR1/NFAT signaling pathway. J Bone Min Res : Off J Am Soc Bone Min Res. 2007;22:1612–20.
Gegenbauer K, Elia G, Blanco-Fernandez A, Smolenski A. Regulator of G-protein signaling 18 integrates activating and inhibitory signaling in platelets. Blood. 2012;119:3799–807.
Delesque-Touchard N, Pendaries C, Volle-Challier C, Millet L, Salel V, Herve C, et al. Regulator of G-protein signaling 18 controls both platelet generation and function. PLoS One. 2014;9, e113215.
Alshbool FZ, Karim ZA, Vemana HP, Conlon C, Lin OA, Khasawneh FT. The regulator of G-protein signaling 18 regulates platelet aggregation, hemostasis and thrombosis. Biochem Biophys Res Commun. 2015;462:378–82.
Lu Q, Sun EE, Klein RS, Flanagan JG. Ephrin-B reverse signaling is mediated by a novel PDZ-RGS protein and selectively inhibits G protein-coupled chemoattraction. Cell. 2001;105:69–79.
Qiu R, Wang J, Tsark W, Lu Q. Essential role of PDZ-RGS3 in the maintenance of neural progenitor cells. Stem Cells. 2010;28:1602–10.
Williams JW, Yau D, Sethakorn N, Kach J, Reed EB, Moore TV, et al. RGS3 controls T lymphocyte migration in a model of Th2-mediated airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2013;305:L693–701.
Mighiu AS, Heximer SP. Controlling parasympathetic regulation of heart rate: a gatekeeper role for RGS proteins in the sinoatrial node. Front Physiol. 2012;3:204.
Han MH, Renthal W, Ring RH, Rahman Z, Psifogeorgou K, Howland D, et al. Brain region specific actions of regulator of G protein signaling 4 oppose morphine reward and dependence but promote analgesia. Biol Psychiatry. 2010;67:761–9.
Stratinaki M, Varidaki A, Mitsi V, Ghose S, Magida J, Dias C, et al. Regulator of G protein signaling 4 [corrected] is a crucial modulator of antidepressant drug action in depression and neuropathic pain models. Proc Natl Acad Sci U S A. 2013;110:8254–9.
Cho H, Park C, Hwang IY, Han SB, Schimel D, Despres D, et al. Rgs5 targeting leads to chronic low blood pressure and a lean body habitus. Mol Cell Biol. 2008;28:2590–7.
Qin M, Huang H, Wang T, Hu H, Liu Y, Cao H, et al. Absence of Rgs5 prolongs cardiac repolarization and predisposes to ventricular tachyarrhythmia in mice. J Mol Cell Cardiol. 2012;53:880–90.
Qin M, Huang H, Wang T, Hu H, Liu Y, Gu Y, et al. Atrial tachyarrhythmia in Rgs5-null mice. PLoS One. 2012;7, e46856.
Cheng WL, Wang PX, Wang T, Zhang Y, Du C, Li H, Ji Y (2014) Regulator of G-protein signalling 5 protects against atherosclerosis in apolipoprotein E-deficient mice. Br J Pharmacol. doi:10.1111/bph.12991.
Takata Y, Liu J, Yin F, Collins AR, Lyon CJ, Lee CH, et al. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci U S A. 2008;105:4277–82.
Kuwata H, Nakao K, Harada T, Matsuda I, Aiba A. Generation of RGS8 null mutant mice by Cre/loxP system. Kobe J Med Sci. 2007;53:275–81.
Saitoh O, Kubo Y, Miyatani Y, Asano T, Nakata H. RGS8 accelerates G-protein-mediated modulation of K+ currents. Nature. 1997;390:525–9.
Benians A, Nobles M, Tinker A. Participation of RGS8 in the ternary complex of agonist, receptor and G-protein. Biochem Soc Trans. 2004;32:1045–7.
Laroche G, Giguere PM, Roth BL, Trejo J, Siderovski DP. RNA interference screen for RGS protein specificity at muscarinic and protease-activated receptors reveals bidirectional modulation of signaling. Am J Physiol Cell Physiol. 2010;299:C654–64.
von Buchholtz L, Elischer A, Tareilus E, Gouka R, Kaiser C, Breer H, et al. RGS21 is a novel regulator of G protein signalling selectively expressed in subpopulations of taste bud cells. Eur J Neurosci. 2004;19:1535–44.
Li X, Chen L, Ji C, Liu B, Gu J, Xu J, et al. Isolation and expression pattern of RGS21 gene, a novel RGS member. Acta Biochim Pol. 2005;52:943–6.
Kimple AJ, Garland AL, Cohen SP, Setola V, Willard FS, Zielinski T, et al. RGS21, a regulator of taste and mucociliary clearance? Laryngoscope. 2014;124:E56–63.
Blazer LL, Storaska AJ, Jutkiewicz EM, Turner EM, Calcagno M, Wade SM, et al. Selectivity and anti-Parkinson’s potential of thiadiazolidinone RGS4 inhibitors. ACS Chem Neurosci. 2015;6:911–9.
Monroy CA, Doorn JA, Roman DL. Modification and functional inhibition of regulator of G-protein signaling 4 (RGS4) by 4-hydroxy-2-nonenal. Chem Res Toxicol. 2013;26:1832–9.
Yoon SY, Woo J, Park JO, Choi EJ, Shin HS, Roh DH, et al. Intrathecal RGS4 inhibitor, CCG50014, reduces nociceptive responses and enhances opioid-mediated analgesic effects in the mouse formalin test. Anesth Analg. 2015;120:671–7.
Lee PC, Sowa ME, Gygi SP, Harper JW. Alternative ubiquitin activation/conjugation cascades interact with N-end rule ubiquitin ligases to control degradation of RGS proteins. Mol Cell. 2011;43:392–405.
Jiang Y, Choi WH, Lee JH, Han DH, Kim JH, Chung YS, et al. A neurostimulant para-chloroamphetamine inhibits the arginylation branch of the N-end rule pathway. Sci Rep. 2014;4:6344.
Raveh A, Schultz PJ, Aschermann L, Carpenter C, Tamayo-Castillo G, Cao S, et al. Identification of protein kinase C activation as a novel mechanism for RGS2 protein upregulation through phenotypic screening of natural product extracts. Mol Pharmacol. 2014;86:406–16.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIAID.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editor: Shelley Hooks
Rights and permissions
About this article

Cite this article
Xie, Z., Chan, E.C. & Druey, K.M. R4 Regulator of G Protein Signaling (RGS) Proteins in Inflammation and Immunity. AAPS J 18, 294–304 (2016). https://doi.org/10.1208/s12248-015-9847-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9847-0