
Abstract
The fact that various immune cells, including macrophages, can be found in tumor tissues has long been known. With the introduction of concept that macrophages differentiate into a classically or alternatively activated phenotype, the role of tumor-associated macrophages (TAMs) is now beginning to be elucidated. TAMs act as “protumoral macrophages”, contributing to disease progression. As the relationship between TAMs and malignant tumors becomes clearer, TAMs are beginning to be seen as potential therapeutic targets in these cases. In this review, we will discuss how TAMs can be used as therapeutic targets of cancer in clinics.
Similar content being viewed by others

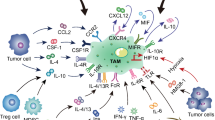
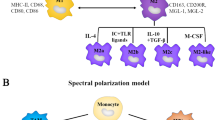
Background
Non-resolving inflammation in a tumor microenvironment is a hallmark of cancer. Leukocytes, fibroblasts, and vascular endothelial cells together form a tumor microenvironment, with immune cells representing its major component. These immune cells interact with tumor cells to influence the initiation, growth, and metastasis of tumors. Tumor-associated macrophages (TAMs), specifically, are often prominent immune cells that orchestrate various factors in the tumor microenvironment [1, 2].
In general, TAMs are thought to more closely resemble M2-polarized macrophages [3], also known as alternatively activated macrophages, which are activated by helper T cell 2 cytokines (e.g., interleukin (IL) -4, IL-10, and IL-13). TAMs play an important role in connecting inflammation with cancer. TAMs can promote proliferation, invasion, and metastasis of tumor cells, stimulate tumor angiogenesis, and inhibit antitumor immune response mediated by T cells, followed by the promotion of tumor progression [3]. There are strong evidences of tumor promotion by TAMs in different cancer models, and an increased TAM prevalence correlates with low survival rates in many human cancers.
With the unraveling of the relationship between TAMs and malignant tumors, TAMs are now being recognized as potential therapeutic targets for cancer. Targeting TAMs is a novel strategy for treatment of cancers. In this review, we summarize how TAMs are used as therapeutic targets in cancers.
Limiting monocyte recruitment
One strategy for targeting TAMs is to block monocyte recruitment into tumor tissues. Targeting the chemokine (C-C motif) ligand 2 (CCL2) - chemokine (C-C motif) receptor (CCR2) axis is promising due to its important role in monocyte recruitment in tumors. A CCL2-blocking agent (carlumab, CNTO88) has been shown to inhibit the growth of several cancers in animal models. A phase II study of carlumab in metastatic castration-resistant prostate cancer patients showed that this antibody was well-tolerated, but that neither blocked the CCL2/CCR2 axis nor showed antitumor activity as a single agent in these metastatic cancer patients [4] (NCT00992186, Table 1). Similar results from Brana et al. showed that carlumab in combination with four chemotherapy regimens for the treatment of patients with solid tumors was well-tolerated, although no long-term suppression of serum CCL2 or significant tumor responses were observed [5] (NCT01204996, Table 1). However, according to the results of other study, carlumab was well-tolerated with evidence of transient CCL2 suppression and preliminary antitumor activity [6] (NCT00537368, Table 1).
Sanford et al. demonstrates that a CCR2 antagonist (PF-04136309) can block the mobilization of CCR2+ monocytes from bone marrow to tumors in a mouse model of pancreatic cancer and can lead to TAM depletion, causing the inhibition of tumor growth and distant metastasis [7]. PF-04136309, in combination with FOLFIRINOX chemotherapy, was used in a phase Ib trial (NCT01413022, Table 1). This therapy was found safe and tolerable with an objective tumor response [8]. Moreover, the efficiency of the humanized antibody specific for CCR2 (MLN1202) was determined in a clinical investigation (NCT01015560, Table 1).
Treatment with systemic CD11b-neutralizing monoclonal antibodies has been shown to prevent the recruitment of myeloid cells to tumors. It has been shown that the use of Mac-1 (CD11b/CD18) antibodies leads to an improved response to radiation therapy in squamous cell carcinoma xenografts of mice, which is accompanied by reduced infiltration of myeloid cells expressing matrix metallopeptidase-9 and S100A8 inside tumors [9].
Because targeting monocytes, prior to being recruited to tumors, has been effective in various cancer models and partial clinical trials, TAMs can be directly targeted as well by other approaches once they invade tumors.
Targeting the activation of TAMs
TAMs can be targeted at the level of activation using various strategies. Colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R) signaling is critical for the generation of monocyte progenitors in bone marrow and TAM polarization in tumor tissues. For these reasons, CSF1/CSF1R signaling is an attractive target for cancer treatment. Genetic loss of CSF1 results in significantly reduced metastasis and delayed tumor progression in breast and neuroendocrine tumor models [10]. Based on these results, several clinical trials of CSF1/CSF1R inhibitors have been completed or are ongoing (Table 1).
Macrophage surface markers can act as useful therapeutic targets. Mannose receptor CD206 can be exploited as a macrophage-specific target. A single-chain peptide bound to the CD206 receptor was attached to nanobodies that can selectively target CD206+ TAMs [11]. Legumain, a stress protein and a member of the asparagine endopeptidase family, can serve as an efficient therapeutic target when overexpressed in TAMs [12]. Targeting surface markers such as scavenger receptor A and CD52 by using immunotoxin-conjugated monoclonal antibodies (mAbs) has been investigated in ovarian cancer [13]. Moreover, the efficiency of alemtuzumab (anti-CD52 antibody) as a tumor treatment in ongoing clinical trials is under investigation (NCT00637390, NCT00073879, Table 1).
Trabectedin (ET743, Yondelis®) was shown to decrease the number of TAMs in tumor tissues by inducing apoptosis of monocytes and macrophages [14]. Based on the favorable results of several phase I, II, and III clinical trials, trabectedin has gained full marketing approval from the European Commission for use in the treatment of ovarian cancer and soft tissue sarcomas and FDA approval in 2015 for use in unresectable or metastatic liposarcoma or leiomyosarcoma [15].
Reprogramming TAMs to antitumor macrophages
As discussed above, one of the key features of macrophages is their plasticity, which enables them to change their phenotype in the tumor microenvironment. Thus, reprogramming TAMs to an antitumor phenotype is an attractive therapeutic strategy. The results of our previous study showed that pseudomonas aeruginosa mannose-sensitive hemagglutinin, which is used in MPE treatment, re-educated CD163+ TAMs to M1 macrophages in MPE, suggesting that reprogramming CD163+ TAMs can be served as a potential therapeutic strategy of MPE [16].
Nanoparticles are gradually used in polarization of TAMs into antitumor macrophages. Recently, Zanganeh et al. found that ferumoxytol significantly inhibited growth of subcutaneous adenocarcinomas in mice, and this tumor growth inhibition was accompanied by an increase in pro-inflammatory M1 macrophages in tumor tissues [17]. Recent data suggest that bioconjugated manganese dioxide nanoparticles enhance the responses of chemotherapy by inducing TAM toward M1-like phenotype [18]. Synthesized nanoparticles with IL-12 payload can reverse macrophages to antitumor function [19].
CD40 is a surface marker of macrophages that can be used to inhibit cytotoxic functions. The combination of a CD40 agonist with gemcitabine in unresectable pancreatic cancer resulted in regression of tumors by promoting antitumor macrophages [20]. ChiLob 7/4 is an intermediate CD40 agonist and chimeric IgG1, which was also shown to induce pro-inflammatory cytokines, with promising results in CD40-expressing solid tumors and diffuse large B-cell lymphoma resistant to conventional therapy in a phase I clinical trial [21] (NCT01561911, Table 1). Other clinical trials of molecules targeting CD40 for cancer treatment are ongoing (NCT01433172, NCT01103635, Table 1).
Activation of the nuclear factor κB pathway also plays an important role in polarization of TAMs to an antitumor phenotype using Toll-like receptor agonists, anti-CD40 mAbs and IL-10 mAbs [22]. In addition, regulation of STAT1 activity is an attractive strategy to induce an antitumor phenotype in macrophages because of the increase production of IL-12 in a murine carcinoma model. A small molecule inhibitor of STAT3 (WP1066) was found to reverse immune tolerance in patients with malignant gliomas and to selectively induce the expression of costimulatory molecules CD80, CD86, and IL-12 on peripheral and tumor-infiltrating macrophages [23]. An investigation of this agent to treat recurrent malignant gliomas and brain metastasis are ongoing (NCT01904123, Table 1).
Thymosin-α is an immunomodulating hormone that can reeducate TAMs into dendritic cells, which participate in antitumor host responses and produce high level of pro-inflammatory cytokines. Nanodelivery of thymosin-α is a feasible approach to increase immune activity in cancer patients. Moreover, several clinical trials have confirmed that thymosin-α prolongs survival in patients with metastatic melanomas and advanced non-small cell lung cancers [24].
β-glucan, a yeast-derived polysaccharide, has been shown to differentiate TAMs into an M1 phenotype, and is a potent immunomodulator with anti-cancer properties. The use of β-glucan is currently under investigation in a phase I clinical trial of patients with neuroblastoma [25] (NCT00911560, Table 1). In another clinical trial, a β-glucan polymer (PGG) showed compelling but modest activity in a phase II multi-cancer study [26] (NCT00912327, Table 1). Furthermore, the efficiency of β-glucan is currently under phase I clinical investigation (NCT00492167, Table 1).
In addition, CD47 has been identified as an important “don’t eat me” signal expressed on malignant cells [27]. Blockade of the CD47:SIRP-α axis between tumor cells and macrophages increases tumor cell phagocytosis in both solid tumors and hematological malignancies. Two phase I dose escalation trials are currently underway with anti-CD47 antibodies as a monotherapy for the treatment of advanced solid tumors and hematological cancers [27] (NCT02216409, NCT02367196, Table 1). CD47 blocking agents are expected to be well tolerated, efficacious and broadly applicable for cancer therapies.
TAMs as a carry of anti-cancer drugs
TAMs can also be used as a carry of anti-cancer drugs, which is one of the most promising strategies of cancer treatment. It has been reported that macrophages can actively internalize gold nanoshells and deliver them into hypoxic regions of tumors [28], inducing cancer cell death around these macrophages. Wang et al. found that macrophages loaded by a magnetic shell combined with topoisomerase I inhibitor SN38 could deliver into the tumor site and exert an anti-cancer effect [29]. In addition, similar combination therapy was showed by Ikehara et al. [30]. Nanoparticles coated with mannose and loaded with 5-fluorouracil were internalized by macrophages. The tumor growth inhibition was observed when an electromagnetic field was applied in a mouse intraperitoneal metastatic model.
Targeting TAMs in combination with standard therapies
Radiotherapy and chemotherapy are useful treatments in many cancers, and studies have shown that infiltrated-myeloid increases after irradiation. However, the interaction between tumor cells and stroma after these therapies remains poorly defined. DNA damage, cell death, and increased hypoxia have been observed in tumors after radiotherapy, which has been shown to lead to macrophage recruitment and promote tumor progression in animal models [31]. Therefore, it is essential to combine TAM targeting with standard therapies for effective tumor treatment.
The hypoxia-inducible factor-1 (HIF-1) pathway is stimulated by radiation-induced tumor hypoxia and the HIF-1 inhibitor can result in decreased infiltration of myeloid cells into tumors [32]. Even more strikingly, blocking CSF1R signaling appears to enhance the efficacy of several other standard therapies. As such, CSF1R blockade has been shown to increase the efficacy of chemotherapy for pancreatic tumors [33].
Conclusions
Targeting TAMs is a promising strategy for cancer treatment. Recent ongoing experimental, pre-clinical, and clinical studies of TAMs have shown encouraging progress. We believe that TAM-targeted therapies would be applied in cancer patients in the future.
Abbreviations
- CCL2:
-
Chemokine (C-C motif) ligand 2
- CCR2:
-
Chemokine (C-C motif) receptor 2
- CSF1:
-
Colony-stimulating factor
- CSF1R:
-
CSF1 receptor
- HIF-1:
-
Hypoxia-inducible factor 1
- IL:
-
Interleukin
- TAMs:
-
Tumor-associated macrophages
References
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–37.
Li L, Yang L, Wang L, Wang F, Zhang Z, Li J, Yue D, Chen X, Ping Y, Huang L, et al. Impaired T cell function in malignant pleural effusion is caused by TGF-β derived predominantly from macrophages. Int J Cancer. 2016;139:2261–9.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Pienta KJ, Machiels JP, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ, Li S, Seetharam S, Puchalski TA, Takimoto C, et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig New Drugs. 2013;31:760–8.
Brana I, Calles A, LoRusso PM, Yee LK, Puchalski TA, Seetharam S, Zhong B, de Boer CJ, Tabernero J, Calvo E. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10:111–23.
Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, Wang G, Tromp BJ, Puchalski TA, Balkwill F, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71:1041–50.
Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, Mitchem JB, Plambeck-Suess SM, Worley LA, Goetz BD, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–15.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17:651–62.
Ahn GO, Tseng D, Liao CH, Dorie MJ, Czechowicz A, Brown JM. Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc Natl Acad Sci U S A. 2010;107:8363–8.
Pyonteck SM, Gadea BB, Wang HW, Gocheva V, Hunter KE, Tang LH, Joyce JA. Deficiency of the macrophage growth factor CSF-1 disrupts pancreatic neuroendocrine tumor development. Oncogene. 2012;31:1459–67.
Movahedi K, Schoonooghe S, Laoui D, Houbracken I, Waelput W, Breckpot K, Bouwens L, Lahoutte T, De Baetselier P, Raes G, et al. Nanobody-based targeting of the macrophage mannose receptor for effective in vivo imaging of tumor-associated macrophages. Cancer Res. 2012;72:4165–77.
Smahel M, Duskova M, Polakova I, Musil J. Enhancement of DNA vaccine potency against legumain. J Immunother. 2014;37:293–303.
Pulaski HL, Spahlinger G, Silva IA, McLean K, Kueck AS, Reynolds RK, Coukos G, Conejo-Garcia JR, Buckanovich RJ. Identifying alemtuzumab as an anti-myeloid cell antiangiogenic therapy for the treatment of ovarian cancer. J Transl Med. 2009;7:49.
Allavena P, Signorelli M, Chieppa M, Erba E, Bianchi G, Marchesi F, Olimpio CO, Bonardi C, Garbi A, Lissoni A, et al. Anti-inflammatory properties of the novel antitumor agent yondelis (trabectedin): inhibition of macrophage differentiation and cytokine production. Cancer Res. 2005;65:2964–71.
Gordon EM, Sankhala KK, Chawla N, Chawla SP. Trabectedin for soft tissue sarcoma: current status and future perspectives. Adv Ther. 2016;33:1055–71.
Yang L, Wang F, Wang L, Huang L, Wang J, Zhang B, Zhang Y. CD163+ tumor-associated macrophage is a prognostic biomarker and is associated with therapeutic effect on malignant pleural effusion of lung cancer patients. Oncotarget. 2015;6:10592–603.
Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, Pajarinen JS, Nejadnik H, Goodman S, Moseley M, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986–94.
Song M, Liu T, Shi C, Zhang X, Chen X. Bioconjugated manganese dioxide nanoparticles enhance chemotherapy response by priming tumor-associated macrophages toward M1-like phenotype and attenuating tumor hypoxia. ACS Nano. 2016;10:633–47.
Wang Y, Lin YX, Qiao SL, An HW, Ma Y, Qiao ZY, Rajapaksha RP, Wang H. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials. 2017;112:153–63.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–6.
Johnson P, Challis R, Chowdhury F, Gao Y, Harvey M, Geldart T, Kerr P, Chan C, Smith A, Steven N, et al. Clinical and biological effects of an agonist anti-CD40 antibody: a Cancer Research UK phase I study. Clin Cancer Res. 2015;21:1321–8.
Seya T, Shime H, Matsumoto M. TAMable tumor-associated macrophages in response to innate RNA sensing. Oncoimmunology. 2012;1:1000–1.
Hussain SF, Kong LY, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB. A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res. 2007;67:9630–6.
Garaci E, Pica F, Serafino A, Balestrieri E, Matteucci C, Moroni G, Sorrentino R, Zonfrillo M, Pierimarchi P, Sinibaldi-Vallebona P. Thymosin alpha1 and cancer: action on immune effector and tumor target cells. Ann N Y Acad Sci. 2012;1269:26–33.
Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clin Cancer Res. 2014;20:1375–82.
Segal NH, Gada P, Senzer N, Gargano MA, Patchen ML, Saltz LB. A phase II efficacy and safety, open-label, multicenter study of Imprime PGG injection in combination with Cetuximab in patients with stage IV KRAS-mutant colorectal cancer. Clin Colorectal Canc. 2016;15:222–7.
McCracken MN, Cha AC, Weissman IL. Molecular pathways: activating T cells after cancer cell Phagocytosis from blockade of CD47 “Don’t eat me” signals. Clin Cancer Res. 2015;21:3597–601.
Choi M-R, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D, Badve S, Sturgis J, Robinson JP, Bashir R, et al. A cellular Trojan horse for delivery of therapeutic nanoparticles into tumors. Nano Lett. 2007;7:3759–65.
Wang H, Shrestha TB, Basel MT, Dani RK, Seo GM, Balivada S, Pyle MM, Prock H, Koper OB, Thapa PS, et al. Magnetic-Fe/Fe(3)O(4)-nanoparticle-bound SN38 as carboxylesterase-cleavable prodrug for the delivery to tumors within monocytes/macrophages. Beilstein J Nanotechnol. 2012;3:444–55.
Ikehara Y, Niwa T, Biao L, Ikehara SK, Ohashi N, Kobayashi T, Shimizu Y, Kojima N, Nakanishi H. A carbohydrate recognition-based drug delivery and controlled release system using intraperitoneal macrophages as a cellular vehicle. Cancer Res. 2006;66:8740–8.
De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23:277–86.
Kioi M, Vogel H, Schultz G, Hoffman RM, Harsh GR, Brown JM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. 2010;120:694–705.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–41.
Acknowledgements
Not applicable.
Authors’ contributors
LY wrote and edited the manuscript, YZ revised the manuscript. All authors read and approved the final manuscript.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No.81171986, No.81602024), Funding from State’s Key Project of Research and Development Plan (No. 2016YFC1303500), International Research Cooperation Grant from Science and Technology Department of Henan Province (No.162102410059), Research Grant from the Ministry of Public Health (No.201501004).
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yang, L., Zhang, Y. Tumor-associated macrophages, potential targets for cancer treatment. Biomark Res 5, 25 (2017). https://doi.org/10.1186/s40364-017-0106-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-017-0106-7