
Abstract
Background
Emergence of cross-resistance to current anti-malarial drugs has led to an urgent need for identification of potential compounds with novel modes of action and anti-malarial activity against the resistant strains. One of the most promising therapeutic targets of anti-malarial agents related to food vacuole of malaria parasite is haemozoin, a product formed by the parasite through haemoglobin degradation.
Methods
With this in mind, this study developed two-dimensional-quantitative structure–activity relationships (QSAR) models of a series of 21 haemozoin inhibitors to explore the useful physicochemical parameters of the active compounds for estimation of anti-malarial activities. The 2D-QSAR model with good statistical quality using partial least square method was generated after removing the outliers.
Results
Five two-dimensional descriptors of the training set were selected: atom count (a_ICM); adjacency and distance matrix descriptor (GCUT_SLOGP_2: the third GCUT descriptor using atomic contribution to logP); average total charge sum (h_pavgQ) in pKa prediction (pH = 7); a very low negative partial charge, including aromatic carbons which have a heteroatom-substitution in “ortho” position (PEOE_VSA-0) and molecular descriptor (rsynth: estimating the synthesizability of molecules as the fraction of heavy atoms that can be traced back to starting material fragments resulting from retrosynthetic rules), respectively. The model suggests that the anti-malarial activity of haemozoin inhibitors increases with molecules that have higher average total charge sum in pKa prediction (pH = 7). QSAR model also highlights that the descriptor using atomic contribution to logP or the distance matrix descriptor (GCUT_SLOGP_2), and structural component of the molecules, including topological descriptors does make for better anti-malarial activity.
Conclusions
The model is capable of predicting the anti-malarial activities of anti-haemozoin compounds. In addition, the selected molecular descriptors in this QSAR model are helpful in designing more efficient compounds against the P. falciparum 3D7A strain.
Similar content being viewed by others

Background
Malaria is a deadly infectious disease with about 228 million infected cases and 405,000 deaths worldwide, as recorded in 2018 [1]. The disease is caused by the bite of a mosquito having the Plasmodium parasite, which consists of five main species, Plasmodium falciparum, Plasmodium vivax, Plasmodium ovale, Plasmodium knowlesi and Plasmodium malariae [2]. Of these species, 90% of deaths (mostly in children) were related to P. falciparum [3]. Anti-malarial drugs, such as quinine, chloroquine, artemisinin, proguanil, pyrimethamine, mefloquine, and atovaquone, have been indicated as malaria treatment [4,5,6]. However, Plasmodium species developed resistance to most of these commonly used drugs. This resistance and the lack of a vaccine has become a major problem in malarial treatment in recent years [7]. Therefore, there is a pressing need to improve the efficiency by modifying existing compounds to face drug-resistance, as well as to discover novel anti-malarial compounds.
Due to funding investment constraints, in silico and collaborative approaches have become particularly attractive approaches for malaria drug discovery efforts. Some in silico techniques, namely molecular docking, pharmacophore models or quantitative structure–activity relationships (QSARs) significantly reduce the time and cost in the drug discovery process. Among the techniques, QSAR is considered a valuable tool that is applied extensively in rational drug design. The predictive QSAR model provides a mathematical correlation between the structural properties of the compounds and their anti-malarial activities using one-, two-, and three-dimensional descriptors of physicochemical properties, as well as structural characteristics relating to the activity. Once a reliable QSAR model has been developed, the biological activities of molecules can be predicted from the molecular descriptors by different methodologies, such as multiple linear regression (MLR), partial least squares (PLS), artificial neural networks (ANN) and heuristic method (HM). In recent years, QSAR models were applied to a variety of anti-malarial compounds to figure out physicochemical and structural characteristics that are essential for their activity [8,9,10,11,12,13]. Some QSAR models developed using sulfonamide and its derivatives, 5-(2-methylbenzimidazol-1-yl)-N-alklythiophene-2-carboxamid derivatives in order to select models that had the best predicting ability [6, 14]. Other studies used three-dimensional QSAR (3D-QSAR) combining with extra analysis gave striking structural characteristics that related to anti-malarial efficacy and the mechanism of action of anti-malarial compounds [15,16,17].
The anti-malarial activities of various groups of compounds, in particular quinine and its derivatives, had a satisfactory correlation with their anti-haemozoin activity [18]. Haemozoin is formed inside the food vacuoles of parasites to prevent lethal toxicity of haem, which is a product of the catabolism of haemoglobin. Thus, anti-haemozoin is an important therapeutic target in anti-malarial treatment. Recently, different approaches were highlighted. These approaches include the high-throughput screening (HTS) of anti-malarial drugs based on their physicochemical properties of haemozoin formation, or building computational models for in silico to screen novel anti-malarial drugs, or analog development from natural compounds or existing agents [19]. Of which, prediction models of correlation between anti-haemozoin and anti-malarial activities strongly assist in anti-malarial drug discovery, from modifying known compounds to identifying new chemical scaffolds for different targets of a large diverse database of compounds [18]. However, there is no QSAR model for anti-malarial activity of anti-haemozoin inhibitors. The aim of this study was to develop quantitative structure–activity relationship models to determine the influences of physiochemical structures of haemozoin inhibitors on anti-malarial activities.
Methods
The best QSAR model will be chosen and could be applied for screening and designing better anti-haemozoin compounds for anti-malarial activities in next studies. QSAR modelling was conducted for anti-malarial activities of haemozoin inhibitors using the multiple linear regression (MLR) and partial least square (PLS) methods. Database of 21 compounds possessing both anti-malarial and anti-haemozoin activities were used for building QSAR models. The IC50 of these compounds varied from 0.06 – 10.5 µM (or pIC50 ranged between -1.02 to 1.22). The QSAR model was chosen based on the predicted fitness plots and statistical values of the models. Evaluation of QSAR models depended on three data sets, the training, validation and test sets. The results included the corresponding descriptors (coefficients) and correlation of the observed—predicted values of anti-malarial activities and the statistical parameters. The parameters, correlation coefficient or coefficient of determination (R2 or r-squared), cross-validated r2 (or Q2) and r2 for the external test set (R2_pred), and root mean square error (RMSE) as fitting criteria, were employed to evaluate the goodness of the models. The predictive model was tested based on different methods, such as internal for training set and external validation for test set, as well as Y-randomization method.
Data set
To perform 2D-QSAR, a complete data set containing 21 anti-haemozoin compounds (Table 1) was taken from the experimental anti-malarial activities identifed in a previous work [20]. The half maximal inhibitory concentration (IC50) of the anti-haemozoin compounds was converted to logarithmic scale (pIC50) and used as the dependent variable. These compounds were randomly divided into two subsets, a training set (16 compounds) and a test set (6 compounds).
2D-QSAR
A flowchart for developing 2D-QSAR was conducted following eight steps (Fig. 1). Initially, database included 21 compounds having anti-Plasmodium 3D7A activity. The IC50 values of these compounds were converted into logarithm scale logIC50 or pIC50 (pIC50 =− logIC50). The process of energy minimization of the compounds was performed using MOE 2015.10. A further step was the calculation of 2D descriptors. A total of 206 descriptors described molecular structures, including geometrical, physicochemical, sterical and lipophilic, which were calculated using Descriptors tool in MOE 2015.10. The database was subsequently divided into two subsets, a training set and a test set, with a 75:25 ratio. The database was divided randomly using RAND or Diverse subset using MOE. Selection of descriptors was carried out carefully. Some descriptors were removed based on three methods, firstly, if more than 15% compounds had descriptor values of 0 using Microsoft Excel. Secondly, using Rapidminer Studio 8.2.0 to take out of descriptors of the compounds which possess 50% similarity. Thirdly, remove randomly one of two descriptors having a cross correlation value of more than 70% using Rapidminer. These selected descriptors were also separated according the ratios of between 0 to 1 using Normalize in Rapidminer Studio based on the Eq. 1 below.
of which: \({\text{X}}_{{\text{n}}}\): Value; \({\text{X}}_{0}\): Initial value; \({\text{Min}}_{0}\),\({\text{ Max}}_{0}\): Minimum, maximum of initial values.

General steps of generating the QSAR model
Contigency tool in MOE and BestFirst—a searching method with assessment algorithm CfsSubsetEval in Weka 3.8.1 were used to find out the suitable descriptors.
Some outliers were removed by using PCA or Z-score, if the compounds had Z-score values of more than 2.0 before building 2D-QSAR. Using MOE with Model tool, 2D-QSAR models were developed using MLR. The best models were selected based on the highest values of the square of the coefficient of determination (R2) value, internally cross-validated R2 (Q2), and the external validated R2 (R2_pred). Of which, external validation used the test set while the training set was for model development. The internal validation parameters that were used, represented models’ goodness-of-fit and robustness. Finaly, evaluation of 2D-QSAR model on two datasets, training set and test set: Internal and external validations were conducted. The internal validation used the leave-one-out (LOO) cross-validation to internally validate the QSAR model. This is done by excluding the point(s) of training set data, then constructing the model based on the remaining data activities and finally, using this model to test the excluded data. This process was repeated until the training set activities were predicted. The coefficient of cross-validated R2 (or Q2) was calculated for the training set. The external validation was using the model for prediction of the biological activities of test set. The value of predicted correlation coefficient (R2_pred) value was calculated for the test set.
Results and discussion
To conduct this study, database of 21 anti-haemozoin compounds was taken for building 2D-QSAR models (Table 1) to explore the structure–activity relationship of haemozoin inhibitors acting as anti-malarial agents. These compounds had in vitro anti-malarial activities against P. falciparum 3D7A and were used for QSAR modelling. The data set was randomly split into a training set (15 compounds) for model construction and test set (6 compounds), for validation of the model, respectively. The quality of a built QSAR model was demonstrated by the fitting and its predicting ability.
Variable selection
Five two-dimensional descriptors of the training set were selected for QSAR modelling as they all had low inter-correlation (Table 2). They included atom count (a_ICM); + adjacency and distance matrix descriptor (GCUT_SLOGP_2: the third GCUT descriptor using atomic contribution to logP (using the Wildman and Crippen SlogP method) instead of partial charge); average total charge sum (h_pavgQ) in pKa prediction (pH = 7); a very low negative partial charge, including aromatic carbons which have a heteroatom-substitution in “ortho” position (PEOE_VSA-0) and molecular descriptor (rsynth: estimating the synthesizability of molecules as the fraction of heavy atoms that can be traced back to starting material fragments resulting from retrosynthetic rules). The study demonstrated that the average total charge sum (h_pavgQ) in pKa prediction (pH = 7) was the most important descriptor with the correlation coefficient values of about 0.41 (Table 2).
QSAR model development
After selecting molecular descriptors, the linear QSAR models were built using the training set data. The outliers were checked and removed based on their values of PCA (principal component analysis), Z-score, and ZX-score of more than 2. There are four QSAR models that were developed based on the selection of different methods, namely PLS (Partial least squares) and PCR (Principal component regression), respectively with or without outliers (Table 3).
Validation of QSAR models
The evaluation of the QSAR models included the internal and external validations. The parameters for internal validation were R2 (a correlation coefficient), Q2 (predictive ability of the built QSAR models in the training set data employing leave-one-out (LOO) cross-validation method), and R2_pred (predictive ability for the test set). QSAR model is selected if it complies with the three criteria: the values of the high correlation coefficient (R2) between the experimental and the predicted values, the predictive ability of the model for the training set Q2 > 0.5, and the low standard deviation (RMSE). The comparison of four generated 2D-QSAR models were evaluated and compared in Table 3. The results showed that the QSAR models gave similar evaluation results by using PLS or PCR methods with outliers. This means that using different methods for the whole training dataset did not affect the development of the QSAR models. However, after removing outliers, the PLS model gave the better results, and the PCR model without outliers was worst than the others (Table 3). Therefore, the best QSAR model was the PLS model without outliers. The regression equation is represented as following:
where: R2 = 0.745031, RMSE = 0.166261, Q2 = 0.316410, and R2_pred = 0.9554.
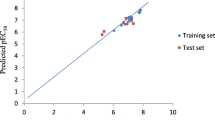
The high values of R2 = 0.745; low standard error (RMSE = 0.166) and the good predictive ability: R2_Pred = 0.9554 (for the test set) indicated suitability of the model for predicting the anti-malarial activities of other haemozoin inhibitors from the existing anti-haemozoin compounds (Table 3). The experimental or observed versus predicted amounts of pIC50 of haemozoin inhibitors as anti-malarial structures against 3D7A strain were presented in Table 4 and Fig. 1. As can be seen in the Table 4, the predicted values of pIC50 values were in good agreement with the values of experimental pIC50.
The linear graphical plot was depicted in Fig. 2. The graph illustrated the good overlap of the observed and predicted activities of the data set with the high of correlation coefficient of R2 = 0.9554 (Fig. 2). The predicted values of pIC50 varied between − 1.021 to 1.222 with the value ranges of the selected descriptors presented in Table 5. The decrease of these descriptors led a decrease of pIC50 values meaning the increase of IC50 which is a decrease of anti-malarial activities.

Plot of the correlation between the experimental pIC50 and the pIC50 predicted anti-malarial activities using partial least squares model
Interpretation of descriptors
It was clearly inferred that the average total charge sum (h_pavgQ) in pKa prediction (pH = 7) contributed the most to the values of pIC50, which could be used as one indicator for predicting anti-malarial activities of other anti-haemozoin agents. The higher average total charge sum (h_pavgQ) in pKa prediction (pH = 7) resulted in increasing values of pIC50, or decreasing of IC50, indicating better anti-malarial activities (Table 4). The positive sign of these descriptors indicated that the larger the value of pIC50, the lower IC50 of the compound. In addition, this feature was also taken for evaluation and prediction of anti-malarial activities for some anti-malarial drugs, such as quinine, pyrimethamine, halofantrine and mefloquine. It was found that the higher their calculated h_pavgQ values, the better anti-malarial activities.
Furthermore, the decrease of distance matrix descriptor (GCUT_SLOGP_2) or the third GCUT descriptor using atomic contribution to logP could lead better anti-malarial activity. The result was compatible with the previous study as this descriptor represents for lipophilicity and low lipophilicity, especially at pH 3, 4, and 5 were significantly related to better anti-malarial activity of anti-haemozoin molecules.
In addition, the positive sign of the PEOE_VSA-0 descriptor, a very low negative partial charge, including aromatic carbons which have a heteroatom-substitution in “ortho” position suggests that increasing in the PEOE_VSA-0 will decrease the inhibitory potency of anti-haemozoin compounds. The increase of atom count (a_ICM), topological descriptors or structural components of the molecules have an effect on the variation of anti-malarial inhibitory activity of the anti-haemozoin compounds. Moreover, molecular descriptor (rsynth: estimating the synthesizability of molecules as the fraction of heavy atoms that can be traced back to starting material fragments resulting from retrosynthetic rules) was the least contributive. In addition, the predicted pIC50 in Table 4 were much different with the experimental pIC50 values for the outliers, especially C1, C2, C12, C16. As a result, removing these outlier compounds from the training set for building QSAR model was essential.
The limitation of this study is the toxicity evaluation. In fact, there is no model predicting both the structure–activity and the structure–toxicity relationships, but they are separate models either predicting the structure–activity or the structure toxicity. Therefore, this QSAR model is not suitable for predicting the toxicity of the compounds. Another QSAR model for toxicity is required.
Conclusion
With the 15 anti-haemozoin compounds, the satistically satisfactory 2D-QSAR model using PLS method was generated after removing the outliers. Five two-dimensional descriptors of the training set were selected: atom count (a_ICM); adjacentcy and distance matrix descriptor (GCUT_SLOGP_2: the third GCUT descriptor using atomic contribution to logP; average total charge sum (h_pavgQ) in pKa prediction (pH = 7); a very low negative partial charge, including aromatic carbons which have a heteroatom-substitution in “ortho” position (PEOE_VSA-0) and molecular descriptor (rsynth: estimating the synthesizability of molecules as the fraction of heavy atoms that can be traced back to starting material fragments resulting from retrosynthetic rules), respectively. The interpretation of the developed model suggests that the anti-malarial activity of haemozoin inhibitors increases with molecules having higher average total charge sum (h_pavgQ) in pKa prediction (pH = 7). The QSAR model also highlights that the descriptor using atomic contribution to logP or the distance matrix descriptor (GCUT_SLOGP_2), and structural component of the molecules, including topological descriptors does make for better anti-malarial activity.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ANN:
-
Artificial neural networks
- HM:
-
Heuristic method
- HTS:
-
High-throughput screening
- LOO:
-
Leave-one-out
- MLR:
-
Multiple linear regression
- PLS:
-
Partial least squares
- QSAR:
-
Quantitative structure–activity relationship
References
WHO. World malaria report 2018. Geneva, World Health Organization, 2018.
Murray MC, Perkins ME. Chemotherapy of malaria. Annu Rep Med Chem. 1996;31:141–50.
Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, et al. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet. 2004;363:1017–24.
Posner GH, Maxwell JP, O’Dowd H, Krasavin M, Xie S, Shapiro TA. Antimalarial sulfide, sulfone, and sulfonamide trioxanes. Bioorg Med Chem. 2000;8:1361–70.
Ryckebusch A, Deprez-Poulain R, Debreu-Fontaine MA, Vandaele R, Mouray E, Grellier P, et al. Synthesis and antimalarial evaluation of new 1,4-bis(3-aminopropyl)piperazine derivatives. Bioorg Med Chem Lett. 2003;13:3783–7.
Agrawal VK, Srivastava R, Khadikar PV. QSAR studies on some antimalarial sulfonamides. Bioorg Med Chem. 2001;9:3287–93.
Sondo P, Derra K, Lefevre T, Diallo-Nakanabo S, Tarnagda Z, Zampa O, et al. Genetically diverse Plasmodium falciparum infections, within-host competition and symptomatic malaria in humans. Sci Rep. 2019;9:127.
Ojha PK, Roy K. The current status of antimalarial drug research with special reference to application of QSAR models. Comb Chem High Throughput Screening. 2015;18:91–128.
Yousefinejad S, Mahboubifar M, Eskandari R. Quantitative structure-activity relationship to predict the anti-malarial activity in a set of new imidazolopiperazines based on artificial neural networks. Malar J. 2019;18:310.
Ojha PK, Kumar V, Roy J, Roy K. Recent advances in quantitative structure-activity relationship models of antimalarial drugs. Expert Opin Drug Discov. 2021:1–37. (online ahead of print)
Joshi N, Hada R, Gupta S, Khan J, Dobrowolski J, Dhar PK, et al. Highly potent anti-malarial activity of benzopyrano(4,3-b)benzopyran derivatives and in silico interaction analysis with putative target Plasmodium falciparum lactate dehydrogenase. J BiomolStruct Dyn. 2021:1–16. (online ahead of print)
Ferreira LT, Borba JVB. QSAR-based virtual screening of natural products database for identification of potent antimalarial hits. Biomolecules. 2021;11:459.
Hadni H, Elhallaoui M. 2D and 3D-QSAR, molecular docking and ADMET properties in silico studies of azaaurones as antimalarial agents. N J Chem. 2020;44:6553–65.
Vyas VK, Parikh H, Ghate M. 3D QSAR studies on 5-(2-methylbenzimidazol-1-yl)-N-alkylthiophene-2-carboxamide derivatives as P. falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitors. Med Chem Res. 2013;22:2235–43.
Cheng F, Shen J, Luo X, Zhu W, Gu J, Ji R, et al. Molecular docking and 3-D-QSAR studies on the possible antimalarial mechanism of artemisinin analogues. Bioorg Med Chem. 2002;10:2883–91.
Katritzky AR, Kulshyn OV, Stoyanova-Slavova I, Dobchev DA, Kuanar M, Fara DC, et al. Antimalarial activity: a QSAR modeling using CODESSA PRO software. Bioorg Med Chem. 2006;14:2333–57.
Cardoso FJ, de Figueiredo AF, da Silva LM, de Miranda RM, de Almeida RC, Pinheiro JC. A study on antimalarial artemisinin derivatives using MEP maps and multivariate QSAR. J Mol Model. 2008;14:39–48.
Tam DNH, Tawfik GM, El-Qushayri AE, Mehyar GM, Istanbuly S, Karimzadeh S, et al. Correlation between anti-malarial and anti-haemozoin activities of anti-malarial compounds. Malar J. 2020;19:298.
Huy NT, Chi PL, Nagai J, Dang TN, Mbanefo EC, Ahmed AM, et al. High-throughput screening and prediction model building for novel hemozoin inhibitors using physicochemical properties. Antimicrob Agents Chemother. 2017;61:e01607-e1616.
Mosaddeque F, Mizukami S, Kamel MG, Teklemichael AA, Dat TV, Mizuta S, et al. Prediction model for antimalarial activities of hemozoin inhibitors by using physicochemical properties. Antimicrob Agents Chemother. 2018;62:e02424-e2517.
Acknowledgements
None.
Funding
This study was funded in part by Vietnam National Foundation for Science and Technology Development (NAFOSTED) (PTVN: under Decision # 239/QĐ-HĐQL-NAFOSTED and Grant #108.05–2017.312). This research was also partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research from AMED under Grant Number JP20am0101086 to provide library compounds (support number 00859). The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
PTVN designed and conducted the experiments, wrote the manuscript. PTVN, TVD, SM, DLHN, FM, SNK, DHBN, OTD, TLV, GLTN, CQD, SM, DNHT, MPT conducted the experiment and wrote the manuscript. NTH and KH supervised and reviewed final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nguyen, P.T.V., Van Dat, T., Mizukami, S. et al. 2D-quantitative structure–activity relationships model using PLS method for anti-malarial activities of anti-haemozoin compounds. Malar J 20, 264 (2021). https://doi.org/10.1186/s12936-021-03775-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-021-03775-2