
Abstract
Background
We evaluated the incidence and spectrum of pathogenic and likely pathogenic variants of cancer susceptibility genes in BRCA1/2 mutation-negative Korean patients with a high risk for hereditary breast cancer using a comprehensive multigene panel that included 35 cancer susceptibility genes.
Methods
Samples from 120 patients who were negative for BRCA1/2 mutations, but had been diagnosed with breast cancer that was likely hereditary, were prospectively evaluated for the prevalence of high-penetrance and moderate-penetrance germline mutations.
Results
Nine patients (7.5%) had at least one pathogenic or likely pathogenic variant. Ten variants were identified in these patients: TP53 in two patients, PALB2 in three patients, BARD1 in two patients, BRIP1 in two patients, and MRE11A in one patient. We also identified 30 types of 139 variants of unknown significance (VUS). High-penetrance germline mutations, including TP53 and PALB2, tended to occur with high frequency in young (< 35 years) breast cancer patients (4/19, 21.1%) than in those diagnosed with breast cancer at ≥35 years of age (1/101, 1.0%; p = 0.003).
Conclusions
These combined results demonstrate that multigene panels offer an alternative strategy for identifying veiled pathogenic and likely pathogenic mutations in breast cancer susceptibility genes.
Similar content being viewed by others


Background
The identification of BRCA1 and BRCA2 germline mutations as predictors of cancer susceptibility significantly improved the diagnosis and prevention of hereditary breast and ovarian cancers (HBOC). Recent advances in genetic testing have enabled the discovery of novel genes that increase the risk of cancer in patients with familial predisposition. Multiple research laboratories have evaluated these cancer-associated mutations in patients who are negative for BRCA1/2 mutations, but still have a high risk of HBOC. These efforts have identified mutations in moderate-risk genes, such as ATM, BRIP1, CHEK2, BARD1, MRE11A, NBN, RAD50, RAD51, and XRCC2, as well as those in high-penetrance genes, including TP53, PTEN, STK11, CDH1, and PALB2, have been reported across diverse ethnic populations [1].
Next generation sequencing (NGS) can provide detailed genetic information via multi-gene panel assays [2]. However, the application of NGS multigene panel test in a clinical setting represents a considerable challenge. It is necessary to not only validate this novel technique, but also to select candidate susceptibility genes. Furthermore, mutations indicative of cancer susceptibility vary across ethnicities; therefore, it is important to understand the clinical and genetic characteristics of multiple susceptibility genes identified by NGS multigene panels in each ethnic population.
In this study, we used comprehensive multigene panels that included 35 known or suspected cancer susceptibility genes to examine BRCA1/2 mutation-negative Korean patients who had clinical features indicative of hereditary breast cancer. We also investigated the feasibility of multigene panel testing for Korean patients, and evaluated potential clinicopathological risk factors related to germline mutations other than BRCA1/2.
Methods
Study population
The study population included 182 Korean BRCA1/2 mutation-negative breast cancer patients with a familial predisposition who were referred to the Cancer Prevention Center, Yonsei Cancer Center, Seoul, Korea between March 1, 2015 and November 11, 2016. Sixty-two patients opted to not participate. Finally, a total of 120 patients were enrolled in the study. Suspected clinical features of hereditary breast cancer were defined as follows: (1) at least one case of breast or ovarian cancer in first- or second-degree relatives; (2) a first diagnosis of breast cancer before age 40; (3) bilateral breast cancer; and (4) co-diagnosis of breast and ovarian cancers in the same patient.
Panel-based mutation analysis
Germline DNA was extracted from the participants’ peripheral blood samples. We used a customized targeted capture sequencing panel (OncoRisk®, Celemics, Seoul, Korea) which included all coding sequences and intron-exon boundaries of the coding exon from 35 cancer predisposition genes (BRCA1, BRCA2, PALB2, BARD1, BRIP1, RAD51C, RAD51D, RAD50, NBN, MRE11A, ATM, CHEK2, TP53, PTEN, APC, BLM, BMPR1A, CDH1, CDK4, CDKN2A, EPCAM, MEN1, MLH1, MSH2, MSH6, MUTYH, PMS2, POLE, PRSS1, RET, SLX4, SMAD4, STK11, VLH, and WT1). Products with each capture reaction were sequenced by 100 base pair paired-end reads on a MiSeq platform (Illumina, San Diego, CA). High-quality sequencing data with an average depth of 500−1000 folds were obtained.
We identified all single base pair substitutions, insertion-deletions, and copy number variants (CNVs) in each gene. Split-read-based detection of large insertions and deletions was conducted using the Pindel and Manta algorithms. CNVs detected by ExomeDepth software [3] were further crosschecked with our custom pipelines, which retrieved base-level depth of coverage for each binary alignment map (BAM) file using SAMtools software (http://samtools.sourceforge.net) and normalized the depths in the same batch (Additional file 1: Figure S1). All likely deleterious mutations were validated by Sanger sequencing, and all possible large rearrangements were confirmed by the multiplex ligation-dependent probe amplification (MLPA) method (Additional file 1: Figure S2).
Genetic variants were classified using a five-tier system following guidelines from the American College of Medical Genetics and Genomics (ACMG) as follows: pathogenic, likely pathogenic, variants of unknown significance (VUS), likely benign, or benign/polymorphism [4]. We used the Sorting Intolerant From Tolerant (SIFT, http://sift.bii.a-star.edu.sg/) and Polymorphism Phenotyping-2 (PolyPhen-2, http://genetics.bwh.harvard.edu/pph2) to generate in silico predictions of several of the identified nonsynonymous variants. Using large rearrangements of exons, pathogenic and likely pathogenic variants were considered as mutations, for consistency with previous studies [5].
Results
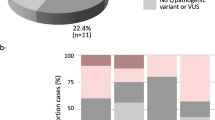
Baseline characteristics of the patients are presented in Additional file 2: Table S1. A total of 7.5% (9/120) of patients were found to carry at least one pathogenic or likely pathogenic variant. A total of ten gene variants (Fig. 1a) were identified in nine patients: TP53 in two patients, PALB2 in three patients, BARD1 in two patients, BRIP1 in two patients, and MRE11A in one patient. We detected a large deletion from exon 2−9 in the TP53 gene, and the other pathogenic variants identified were as follows: PALB2 (c.3267_3268delGT, p.Phe1090SerfsTer6, rs587781890; c.2257C > T, p.Arg753Ter, rs180177110; and c.695delC, p.Gly232ValfsTer6); BARD1 (c.1345C > T, p.Gln449Ter); BRIP1 (c.1066C > T, p.Arg356Ter, rs730881633; and exon 5–6 deletion); and MRE11A (c.1773_1774delAA, p.Gly593LysfsTer4). Likely pathogenic variants were found in TP53 (c.733G > A, p.Gly245Ser, rs28934575). Pathogenic variants in PALB2 and MRE11A were identified in a 34-year-old patient who was co-diagnosed with breast and gastric cancer (Table 1). Three of the pathogenic variants identified in this study were not reported previously.

a Percentage of patients with pathogenic or likely pathogenic mutations corresponding with each gene. b Number of patients with variants of uncertain significance (VUS) for each gene (n = 120 patients total)
A total of 87 patients (72.5%) had at least one VUS (median, 1; range, 0–3). A total of 139 VUS were identified in 30 cancer susceptibility genes, including SLX4 (n = 11), BLM (n = 10), POLE (n = 10), ATM (n = 9), CDH1 (n = 9), CHEK2 (n = 9), BRCA2 (n = 8), RAD50 (n = 7), BRIP1 (n = 6), EPCAM (n = 5), PALB2 (n = 5), PRSS1 (n = 5), TP53 (n = 5), APC (n = 4), MLH1 (n = 4), RET (n = 4), MRE11A (n = 3), MSH2 (n = 3), MSH6 (n = 3), MUTYH (n = 3), RAD51D (n = 3), STK11 (n = 3), BMPR1A (n = 2), BRCA1 (n = 2), CDKN2A (n = 1), MEN1 (n = 1), NBN (n = 1), PMS2 (n = 1), VHL (n = 1), and WT1 (n = 1) (Fig. 1b).
First diagnosis of breast cancer at a relatively young age (<35 years) was correlated with pathogenic or likely-pathogenic variants in high-penetrance cancer susceptibility genes. Pathogenic variants in high-penetrance genes were detected in 21.1% (4/19) of these patients, which was significantly higher than that for patients who were first diagnosed with breast cancer at age ≥ 35 years (1/101, 1.0%, p = 0.003) (Table 2).
Discussion
Previous studies using multigene panel tests identified cancer susceptibility genes in 2.1−16.8% of BRCA1/2 mutation-negative patients [5,6,7,8,9,10,11]. Our tests of high-penetrance genes identified a large exon deletion in TP53, and pathogenic and likely pathogenic variants in TP53 and PALB2 (Table 1). We also identified a frameshift mutation of MRE11A c.1773_1774delAA (p.Gly593LysfsTer4) in a patient with a PALB2 mutation. The MRE11 protein functions in non-homologous end-joining and homologous recombination, which occur during the repair of double-stranded DNA breaks [12]. Therefore, the risk for patients with concurrent dysfunction in PALB2 and MRE11A is unclear and should be assessed in future studies. Because the two frameshift variants in PALB2 (c.3267_3268delGT, p.Phe1090SerfsTer6, rs587781890; and c.695delG, p.Gly232ValfsTer6) were not found in the control group, the variants met the criteria to be likely pathogenic according to the ACMG guideline (PVS1 and PM2) (Table 1) [4]. One nonsense variant in PALB2 (c.2257C > T p.Arg753Ter, rs180177110) had a higher prevalence in affected patients compared to the control group [odds ratio (OR), 127.0; 95% confidence interval (CI), 14.1–1140.1; p < 0.0001]. Therefore, this variant conformed to the criteria to be classified as pathogenic according to ACMG guidelines (PVS1 and PS4) (Table 1) [4]. In addition, a missense variant in TP53, c.733G > A (p.Gly245Ser, rs28934575) was classified as a pathogenic or likely pathogenic variant in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/), and met the criteria for a likely pathogenic variant according to the ACMG guidelines (PM2, PM5, PP2, PP3, and PP5) (Additional file 2: Table S2) [4].
Pathogenic or likely pathogenic variants also were detected in BRCA1-associated RING domain 1 (BARD1) and BRCA1-interacting protein C-terminal helicase 1 (BRIP1). BARD1 and BRIP1 encode proteins that interact with the BRCA1 protein during the repair of DNA double- stranded break, and pathogenic variants of these genes have been investigated [13]. However, there is a controversy as to whether these rare variants are clinically associated with a risk of breast cancer [11, 14]. In a previous study that screened for BRIP1 mutations among 235 Korean patients with BRCA1/2 mutation-negative high-risk breast cancers using fluorescent-conformation sensitive gel electrophoresis (F-CSGE), there was no case of a protein-truncating BRIP1 mutation, which suggests that the prevalence of BRIP1 mutations is likely to be low in the Korean population [15].
Cell cycle checkpoint kinase 2 (CHEK2) is a well-established moderate-penetrance breast cancer gene. Several studies have shown that essentially no case of CHEK2 (c.1100delC) was observed in Asian populations, in contrast to the observed prevalence in European populations [16,17,18,19]. Liu and colleagues reported that the CHEK2 c.1111C > T (p.His371Tyr, rs531398630) variant was observed in 4.24% (5/118) of Chinese familial breast cancer cases without BRCA1/2 mutations, and was associated with dysfunctional phosphorylation of T68 in the SQ/TQ rich domain, which is an activation point following DNA damage [18]. We also identified CHEK2 c.1111C > T variants in 2.5% (3/120) of Korean breast cancer patients without BRCA1/2 mutations (Additional file 2: Table S2). Population-based investigations are required to establish the prevalence of this variant, especially in Asian patients. We identified the CHEK2 c.908 + 2delT variant in one patient, and it was classified as likely pathogenic according to the ACMG guideline (Additional file 2: Table S2). However, we did not classify this variant as a positive result because the experimental study was not sufficient.
In the current study, clinically important likely pathogenic or pathogenic variants of high-penetrance genes were identified in only five (4.2%) patients (TP53 in two patients, and PALB2 in three patients). These variants were identified in 4 of 19 patients (21.1%) with early-onset breast cancer (< 35 years old at onset) (Table 2). A previous study identified cancer susceptibility mutations in 11% of BRCA1/2-negative patients with early-onset breast cancer (diagnosed at <40 years of age) [20]. Considering the frequency of pathogenic variants of high-penetrance genes in patients with early-onset cancer, clinicians should be encouraged to consider performing multigene panel tests for these patients if their conventional BRCA1/2 tests are negative.
This study has several limitations. The primary limitation is the small number of patients (n = 120), which provides only limited data for cancer susceptibility genes in Korean patients with breast cancer. A large-scale cohort study will be required to establish the accurate prevalence and spectrum of pathogenic variants in these patients. The majority of patients (87 of the 120, 72.5%) had VUS. A functional and population-based study will be necessary to clarify the clinical meaning of these VUS. Despite these limitations, to the best of our knowledge, this is the first prospective study to apply customized multigene panels to BRCA1/2 mutation-negative Korean patients with a high risk for HBOC. A recent study conducted by Couch et al. assessed the commercial multigene panel test results of 65,057 patients with breast cancer; however, the frequency, phenotypic association, and cancer risks related to each variant were analyzed among Caucasian women only [11]. Regarding diversity of prevalence of the genetic variants, more prospective studies will be required among diverse ethnic populations.
Conclusions
Wider application of multigene panel tests that include high-penetrance cancer susceptibility genes, so-called “beyond BRCA1/2 genes”, will likely provide clinically relevant information for some patients with high risk for hereditary cancer [1, 13, 21]. However, these panels can produce abundant and conflicting results in clinical practice. To efficiently utilize these data, clinical databases should be established with respect to ethnic backgrounds, and genetic results should be carefully applied for high-risk patients.
Abbreviations
- ACMG:
-
American College of Medical Genetics and Genomics
- AJCC:
-
American Joint Committee on Cancer
- BAM:
-
Binary alignment map
- BARD1:
-
BRCA1-associated RING domain 1
- BRIP1:
-
BRCA1-interacting protein C-terminal helicase 1
- CHEK2:
-
Cell cycle checkpoint kinase 2
- CI:
-
Confidence interval
- CNV:
-
Copy number variants
- ExAC:
-
Exome Aggregation Consortium
- F-CSGE:
-
Fluorescent-conformation sensitive gel electrophoresis
- HBOC:
-
Hereditary breast and ovarian cancers
- MLPA:
-
Multiplex ligation-dependent probe amplification
- NGS:
-
Next generation sequencing
- OR:
-
Odds ratio
- PM:
-
Pathogenic criterion weighted as moderate
- PP:
-
Pathogenic criterion weighted as supporting
- PVS:
-
Pathogenic criterion weighted as very strong
- SAM:
-
Sequence alignment map
- VUS:
-
Variants of unknown significance
References
Young EL, Feng BJ, Stark AW, Damiola F, Durand G, Forey N, Francy TC, Gammon A, Kohlmann WK, Kaphingst KA, et al. Multigene testing of moderate-risk genes: be mindful of the missense. J Med Genet. 2016;53(6):366–76.
Park HS, Park JS, Nam EJ, Lee S-T, Han JW, Kim TI. Clinical implications of genetic testing for hereditary breast and ovarian cancer syndrome in the era of genomic medicine: Clinician’s perspectives. J Breast Dis. 2016;4(1):1–9.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO, Thrasher AJ, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28(21):2747–54.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Susswein LR, Marshall ML, Nusbaum R, Vogel Postula KJ, Weissman SM, Yackowski L, Vaccari EM, Bissonnette J, Booker JK, Cremona ML, et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet Med. 2016;18(8):823–32.
Hirotsu Y, Nakagomi H, Sakamoto I, Amemiya K, Oyama T, Mochizuki H, Omata M. Multigene panel analysis identified germline mutations of DNA repair genes in breast and ovarian cancer. Mol Genet Genomic Med. 2015;3(5):459–66.
Kurian AW, Hare EE, Mills MA, Kingham KE, McPherson L, Whittemore AS, McGuire V, Ladabaum U, Kobayashi Y, Lincoln SE, et al. Clinical evaluation of a multiple-gene sequencing panel for hereditary cancer risk assessment. J Clin Oncol. 2014;32(19):2001–9.
Tung N, Battelli C, Allen B, Kaldate R, Bhatnagar S, Bowles K, Timms K, Garber JE, Herold C, Ellisen L, et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121(1):25–33.
Wong ES, Shekar S, Met-Domestici M, Chan C, Sze M, Yap YS, Rozen SG, Tan M-H, Ang P, Ngeow J. Inherited breast cancer predisposition in Asians: multigene panel testing outcomes from Singapore. NPJ Genomic Medicine. 2016;1:15003.
Crawford B, Adams SB, Sittler T, van den Akker J, Chan S, Leitner O, Ryan L, Gil E, van 't Veer L: Multi-gene panel testing for hereditary cancer predisposition in unsolved high-risk breast and ovarian cancer patients. Breast Cancer Res Treat 2017, 163(2):383–390.
Couch FJ, Shimelis H, Hu C, Hart SN, Polley EC, Na J, Hallberg E, Moore R, Thomas A, Lilyquist J, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;
Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct Mol Biol. 2009;16(8):814–8.
Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat Rev. 2015;41(1):1–8.
Easton DF, Lesueur F, Decker B, Michailidou K, Li J, Allen J, Luccarini C, Pooley KA, Shah M, Bolla MK, et al. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: implications for gene panel testing. J Med Genet. 2016;53(5):298–309.
Kim H, Cho DY, Choi DH, Jung GH, Shin I, Park W, Huh SJ, Nam SJ, Lee JE, Gil WH, et al. Analysis of BRIP1 variants among Korean patients with BRCA1/2 mutation-negative high-risk breast cancer. Cancer Res Treat. 2016;48(3):955–61.
Chen W, Yurong S, Liansheng N. Breast cancer low-penetrance allele 1100delC in the CHEK2 gene: not present in the Chinese familial breast cancer population. Adv Ther. 2008;25(5):496–501.
Choi DH, Cho DY, Lee MH, Park HS, Ahn SH, Son BH, Haffty BG. The CHEK2 1100delC mutation is not present in Korean patients with breast cancer cases tested for BRCA1 and BRCA2 mutation. Breast Cancer Res Treat. 2008;112(3):569–73.
Liu Y, Liao J, Xu Y, Chen W, Liu D, Ouyang T, Li J, Wang T, Fan Z, Fan T, et al. A recurrent CHEK2 p.H371Y mutation is associated with breast cancer risk in Chinese women. Hum Mutat. 2011;32(9):1000–3.
Zhang S, Phelan CM, Zhang P, Rousseau F, Ghadirian P, Robidoux A, Foulkes W, Hamel N, McCready D, Trudeau M, et al. Frequency of the CHEK2 1100delC mutation among women with breast cancer: an international study. Cancer Res. 2008;68(7):2154–7.
Maxwell KN, Wubbenhorst B, D'Andrea K, Garman B, Long JM, Powers J, Rathbun K, Stopfer JE, Zhu J, Bradbury AR, et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med. 2015;17(8):630–8.
Easton DF, Pharoah PD, Antoniou AC, Tischkowitz M, Tavtigian SV, Nathanson KL, Devilee P, Meindl A, Couch FJ, Southey M, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med. 2015;372(23):2243–57.
Park H, Park J, Nam E, Han J, Kim J, Kim T, Cho Y. The prevalence of mutations of breast cancer susceptibility genes in Korean BRCA1/2 mutation-negative patients with a high risk for hereditary breast cancer. Breast. 2017;32:S130.
Monti P, Ciribilli Y, Jordan J, Menichini P, Umbach DM, Resnick MA, Luzzatto L, Inga A, Fronza G. Transcriptional functionality of germ line p53 mutants influences cancer phenotype. Clin Cancer Res. 2007;13(13):3789–95.
Acknowledgements
We are very grateful for the participation in this study of patients and staffs from Breast Cancer Center and Cancer Prevention Center at Yonsei Cancer Center, Severance Hospital, Yonsei University College of Medicine, Seoul, Republic of Korea. The results of this study were presented as a poster at the 15th St.Gallen International Breast Cancer Conference 2017 held on March 15th-18th, 2017, Vienna, Austria [22]. This manuscript includes the abstract presented at the 15th St.Gallen International Breast Cancer Conference 2017.
Funding
This research was supported by the Korea Breast Cancer Foundation (KBCF-2015E002) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2016R1D1A1B03934564).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
JSP designed this study, reviewed the medical records, and wrote the draft. SL and JK carried out NGS, analyzed the data, and interpreted the genetic variant using ACMG guidelines. EJN and JWH discussed the interpretation of data, and critically revised the draft. JL discussed the application of genetic data to the clinic, and critically revised the draft. TIK provided important ideas for analyzing the variant, and coordinated the work of the hereditary cancer clinic. HSP designed this study, wrote the draft, and reviewed the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The prospective study was approved by institutional review board at Severance Hospital, Seoul, Korea (IRB approval number 4–2015-0819). We obtained informed consent in writing from all patients who participated in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Figures S1 and S2.
This file includes the methods detecting pathogenic variants and lage deletion in this study; depth of coverage and method for detection of large insertion-deletion of exon using next-generation sequencing, and confirmation of deleterious mutations using Sanger sequencing or MLPA in four patients. (PDF 1477 kb)
Additional file 2: Tables S1 and S2.
This file includes two tables regarding baseline characteristics of study participants, possibly pathogenic variants and the classification according to ACMG guidelines mentied in the main manuscript. (DOCX 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Park, J.S., Lee, ST., Nam, E.J. et al. Variants of cancer susceptibility genes in Korean BRCA1/2 mutation-negative patients with high risk for hereditary breast cancer. BMC Cancer 18, 83 (2018). https://doi.org/10.1186/s12885-017-3940-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-017-3940-y