
Abstract
Background
Whole-genome sequencing has shown that the Mycobacterium tuberculosis infection process can be more heterogeneous than previously thought. Compartmentalized infections, exogenous reinfections, and microevolution are manifestations of this clonal complexity. The analysis of the mechanisms causing the microevolution —the genetic variability of M. tuberculosis at short time scales— of a parental strain into clonal variants with a patient is a relevant issue that has not been yet completely addressed. To our knowledge, a whole genome sequence microevolution analysis in a single patient with inadequate adherence to treatment has not been previously reported.
Case presentation
In this work, we applied whole genome sequencing analysis for a more in-depth analysis of the microevolution of a parental Mycobacterium tuberculosis strain into clonal variants within a patient with poor treatment compliance in Argentina. We analyzed the whole-genome sequence of 8 consecutive Mycobacterium tuberculosis isolates obtained from a patient within 57-months of intermittent therapy. Nineteen mutations (9 short-term, 10 fixed variants) emerged, most of them associated with drug resistance. The first isolate was already resistant to isoniazid, rifampicin, and streptomycin, thereafter the strain developed resistance to fluoroquinolones and pyrazinamide. Surprisingly, isolates remained susceptible to the pro-drug ethionamide after acquiring a frameshift mutation in ethA, a gene required for its activation. We also found a novel variant, (T-54G), in the 5′ untranslated region of whiB7 (T-54G), a region allegedly related to kanamycin resistance. Notably, discrepancies between canonical and phage-based susceptibility testing to kanamycin were previously found for the isolate harboring this mutation. In our patient, microevolution was mainly driven by drug selective pressure. Rare short-term mutations fixed together with resistance-conferring mutations during therapy.
Conclusions
This report highlights the relevance of whole-genome sequencing analysis in the clinic for characterization of pre-XDR and MDR resistance profile, particularly in patients with incomplete and/or intermittent treatment.
Similar content being viewed by others
Background
Tuberculosis (TB) ranks among the top 10 infectious killers in the world and multidrug-resistant TB (MDR-TB) − TB caused by bacilli resistant to the key first-line anti-TB drugs, isoniazid (INH) and rifampicin (RIF) – is largely responsible for TB mortality [1]. Mycobacterium tuberculosis, the causative agent, is an obligate human pathogen that has coevolved with its host and lacks inter-genomic mobility. Thus, unlike other bacterial species, it does not acquire drug resistance by horizontal gene transfer. Instead, it undergoes a stepwise process triggered by the spontaneous emergence of drug-resistant clones followed by fixation of resistant mutants in the bacillary population; transmission of the resistant clones perpetuates them in the community [2]. The primary mechanism underlying M. tuberculosis drug resistance is the acquisition of mutations in drug-targets or genes coding drug activating enzymes [3]. Recent work showed that the frequency of mutations causing drug resistance is boosted during drug exposure of the bacilli [4].
The treatment success of MDR-TB relies on case detection, drug susceptibility testing (DST) availability, access to second-line drugs, and prompt initiation of adequate therapy. Traditionally, a history of prior treatment for TB has been deemed as the most common risk factor for MDR-TB. However, growing genotyping evidence indicates that the worldwide MDR-TB epidemic is mainly driven by direct transmission of MDR M. tuberculosis strains [5]. Besides, under-recognized and under-treated MDR-TB patients maintain drug-resistant strains in the community for prolonged periods through uninterrupted chains of transmission, thus increasing the risk of spontaneous emergence of additional drug-resistance conferring mutations [6].
Genotyping methods − such as mycobacterial interspersed repetitive units-variable number tandem repeat (MIRU-VNTR) and spoligotyping − have disclosed the existence of a wide intra-species genomic diversity in M. tuberculosis. These methodologies made it possible to distinguish relapses from reinfections and mixed infections concurring within a single host. More recently, the introduction of whole-genome sequencing (WGS) has changed the way we think about M. tuberculosis population dynamics and epidemiology. Nowadays, it is possible to get a deep insight into the timeline of drug resistance acquisition [7, 8], decipher how the transmission events occur, and integrate all the data into a phylogeographic setting [9]. Furthermore, WGS shows that the M. tuberculosis infection process can be more heterogeneous than previously thought. Compartmentalized infections, exogenous reinfections, and microevolution processes (i.e. genetic variability in short-time scale) are different versions of this clonal complexity [7, 10]. In particular, the microevolution of a parental M. tuberculosis strain into clonal variants within a single patient is a relevant issue to be addressed.
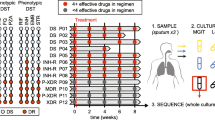
Argentina is a South American country with a moderate burden of TB and disparate rates across the community. Vulnerable groups, such as migrants from neighboring countries, prisoners and previously-treated active cases of MDR-TB and XDR-TB (extensively drug-resistant tuberculosis, i.e. MDR-TB with additional resistance to at least one fluoroquinolone and 1 sec-line injectable drug) are still posing a significant challenge [11, 12]. This case study explores the genomic diversity of M. tuberculosis in an Argentinean patient with a history of TB and inadequate adherence to treatment. To understand the evolution of the TB infection in this patient, we performed WGS of 8-time serial isolates obtained during a 57-month period. We report results on the phylogenetic analyses, microevolution, and relation between genomic variants and phenotypic resistance.
Case presentation
Patient clinical records
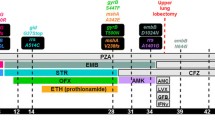
A 40-year-old HIV-negative woman – who was born in the northwestern region of Argentina and worked as a cleaner in a private health center in Buenos Aires − was diagnosed with pulmonary TB in 2008, based on clinical and radiological data (multiple cavitary lesions) together with the presence of acid-fast bacilli (AFB) in sputum smear examination at Argerich Hospital. The patient started standard anti-TB treatment consisting of 2 months RIF, INH, pyrazinamide (PZA), and ethambutol (EMB), followed by 4 months RIF and INH (Fig. 1a). No data about the tuberculosis status could be obtained at the end of this treatment. In 2009, she returned to Argerich Hospital with AFB-positive sputum and lung cavitation. The treatment started again with RIF, INH, PZA, and EMB plus levofloxacin (LVX) for 10 months. In 2010, the patient became AFB smear-negative and remained so for about a year, denoting that the patient was cured. In August 2011, she relapsed with AFB smear-positive TB. At this point, the first isolate was obtained (TB1) at Muñiz Hospital, showing M. tuberculosis resistant to INH, RIF and STR, susceptible to kanamycin (KAN), amikacin (AMK), ofloxacin (OFX), and EMB. LVX minimal inhibitory concentration (MIC) was < 0.5 mg/L. A new drug combination scheme was started, consisting of terizidone, para-aminosalicylic acid (PAS), ethionamide (ETH), OFX, and KAN for 9 months. Three months later, the patient remained AFB smear-positive and culture-positive (TB2), with a DST profile identical to the first one. In March 2012 (TB3, TB4), the patient was still bacteriologically positive, and the isolate showed additional resistance to OFX and LVX (MIC 12 μg/ml). The isolate remained susceptible to PZA, EMB, KAN, AMK, capreomycin (CP), ETH, cycloserine (Cs), PAS, and moxifloxacin (MFX). Based on these results, a new therapy was established with PZA, EMB, KAN, ETH, Cs, PAS, and linezolid (LNZ). In July 2012, the patient persisted AFB smear-positive and was hospitalized in Muñiz Hospital. In December (TB5), the isolate was resistant to INH, RIF, STR, OFX, and LVX and persisted susceptible to KAN, AMK, CP, PZA, PAS, Cs, ETH, LNZ, and MFX.

Patient history and clinical isolates. a, Patient history and clinical records. b, Phylogeny of 8 time-serial isolates with drug-resistance. Resistance-conferring mutations are directly indicated on the tree. TERI: terizidone, PAS: para-aminosalicylic acid, OFX: ofloxacin, KAN: kanamycin, PZA: pyrazinamide, Cs: cycloserine, LNZ: linezolid, MXF: moxifloxacin, MXF-hd: moxifloxacin high dose, IC: imipenem-clavulanate, MC: meropenem-clavulanate AMK: amikacin. STR: streptomycin, INH: isoniazid, RIF: rifampicin, ETH: ethionamide, CFZ: clofazimine, AMGs: aminoglycosides
In September 2013 (TB6), the patient was hospitalized again and was prescribed a second-line treatment scheme with MXF, PAS, Cs, KAN, PZA, and ETH. The ETH was taken irregularly due to intolerance until it was suspended. After 7 months, the patient was discharged from hospital. As an outpatient, she did not adhere properly to therapy.
In 2015 (TB7), the patient returned symptomatic and was readmitted to Muñiz Hospital where she was treated empirically with MXF, PAS, Cs, KAN, PZA, ETH, LZN, and meropenem-clavulanate while waiting for results of a new DST. Once again, the ETH had to be suspended due to intolerance. The DST results showed resistance to INH, RIF, STR, OFX, LVX, PZA and MXF, this latter at 0.5 μg/ml. In view of these results, the drug scheme was adjusted to high-dose MXF, Cs, PAS, KAN, meropenem-clavulanate, and LNZ. After 10 months, this treatment was discontinued due to a supply shortage in the hospital pharmacy. In May 2016 (TB8), the therapy was resumed, but meropenem-clavulanate was replaced by imipenem-clavulanate and AMK was administered instead of KAN. This drug scheme was accomplished under supervision for 7 months. After 152 days of AMK and 209 days of imipenem-clavulanate with favourable evolution, the patient was discharged and injectable drugs were discontinued. She continued treatment as an outpatient under regular controls. By the end of 2016, the patient remained asymptomatic and negative to AFB smear and culture, denoting the patient was completely cured (Fig. 1a).
Clinical isolates
We analyzed the 8 available isolates obtained from the same patient between August 2011 and January 2016 at Hospital Muñiz. Identification and DST to first- and second-line anti-TB drugs was performed at Muñiz Hospital and confirmed at the TB National Reference Laboratory (NRL) at the National Institute of Infectious Diseases Dr. Carlos Malbrán (ANLIS). The first 4 primary sputum cultures (TB1 to TB4) were collected at Argerich Hospital, the rest (TB5 to TB8) were collected at Hospital Muñiz; both hospitals are located in Buenos Aires. A previous TB episode occurred in 2008, and by 2010 the patient had become smear-negative and was discharged. Unfortunately, no isolate was available from that first episode, and we cannot determine if the following TB episode in August 2011 was caused by the same strain.
Microbiological and molecular studies
All isolates were grown on Löwenstein-Jensen slants and identified as M. tuberculosis by biochemical and molecular tests. DST was performed by the reference standard proportion method in Löwenstein-Jensen medium and/or BACTEC MGIT 960 (Becton Dickinson, MD) under international standards [13]. A multiplex allele-specific PCR (MAS-PCR) for detection of mutations conferring resistance to INH and RIF (codons katG315, inhA-15, rpoB450, 445, and 425) was performed on all isolates according to a modified protocol described elsewhere [14].
Genotyping was performed by spoligotyping and MIRU-VNTR according to standard procedures [15, 16], followed by comparison with SITVITWEB [17] and MIRU-VNTRplus database [18].
Genome sequencing
To perform WGS, we re-cultured the isolates on Löwenstein-Jensen slants. The DNA was extracted according to a standard protocol for mycobacteria [19]. Genomic libraries were constructed using Nextera® XT DNA Sample Preparation Kit (Illumina) following the manufacturer’s instructions. Individual libraries were indexed with Nextera® XT Index Kit. Paired-end reads from all the isolates were obtained using the Illumina MiSeq platform at ANLIS. All reads were deposited in SRA NCBI, Accession number PRJNA646920.
Variant calling
Raw reads were checked for quality using FastQC version v0.11.5 [20] and processed with the PRINSEQ lite version 0.20.4 (reads shorter than 36 bp and mean Q < 20 were filtered out) [20, 21] ensuring quality assessment. The reads were aligned to the H37Rv reference genome (NCBI access number NC_000962.3) using BWA 0.7.17 [22]. Variant calling was made with GATK 4.1.8.0 [23]. After filtering problematic regions, we applied 3 different approaches to the analysis: first, we called variants at a low frequency (0.25) to explore short-term mutations, then we called variants at a higher frequency (0.75) to infer phylogenetic trees. Finally, we called even more frequent variants (0.9) to identify fixed mutations within bacillary populations. The resulting variants were annotated with SnpEff 4_3t [24] and compared with our database of drug resistance-associated variants which combines data of TB Profiler v2.8.8 [25], KvarQ v0.12.2 [26], CARD RGI 5.1.1 (database downloaded on June 18, 2020) [27], and bibliographic data. The analysis of the variant-containing genes was performed with Target Pathogen [28].
Phylogenetic analyses
The phylogenetic analysis was performed by comparing the samples TB1 to TB8 with representative samples of the main M. tuberculosis lineages and sublineages [29] downloaded from NCBI. The reads were processed using the variant calling pipeline mentioned above. The resulting variants from all samples were combined in a single VCF using GATK. To confirm that all the patient’s isolates composed a single monophyletic clade, we constructed a maximum-likelihood genome phylogenetic tree using RaXML v8.2.12 [30] dataset used includes 1 or more representative genomes for each M. tuberculosis sublineage.
Nineteen mutations emerged among the 8 time-serial isolates over 57-month of intermittent therapy. Nine out of these 19 variants reached fixation in the bacilli population. The other 10 were categorized as short-term mutations, i.e. single nucleotide polymorphism (SNPs) and Insertion–deletion mutations (InDels) that arise and fade-out within months or years (Supplementary Table 1). The most recent common ancestor was MDR and evolved to a pre-XDR status within 7 months (Fig. 1b).
The clonal nature of the isolates was supported by identical MIRUs-VNTR and spoligotyping patterns (Fig. 2a). Spoligotyping results showed that all the samples belonged to SIT450, an ambiguous clade between X1 and T5. These sublineages belong to the 4 Euro-American lineage, which is the predominant in Latin America [32].

a. MIRUs-VNTR and spoligotype patterns displayed by the eight time-serial isolates. b. Phylogenetic reconstruction based on the eight time-serial isolates analyzed and 193 genomes representative of all M. tuberculosis lineages and sub-lineages according to SNP barcode classification by Coll et al. [31]
To place the patient’s isolates in a global phylogenetic context, and confirm they form a single monophyletic clade, we constructed a maximum-likelihood core genome phylogenetic tree. According to a recent SNP-based phylogenetic classification [31], the 8 isolates composed a monophyletic cluster assigned to the sub-lineage 4.1.1, which corresponds to the X type (Fig. 2b).
Drug-resistance conferring mutations and putative compensatory mutations
In August 2011, the TB NRL received the first isolate (TB1), and 3 months later the second one (TB2). Both isolates were found to be already resistant to INH and RIF – that is, they qualified as MDR – with additional resistance to STR. These isolates harbored katG S315T, rpoB D435V, and rrs C517T, mutations related to resistance to these three drugs, respectively. These 3 mutations remained fixed in all 8 isolates until the end of the treatment. The genotypic prediction of resistance from WGS data was in line with the results of the phenotypic and molecular techniques applied at the TB NRL (Table 1 and Supplementary Table 2).
By March 2012 (TB3), phenotypic resistance to OFX and LVX was detected, with borderline resistance to MXF, which changed the strain status to pre-XDR. This phenotypic resistance was supported by the mutation gyrA D94H. The strain remained phenotypically resistant to fluoroquinolones until the end, but the frequency of this mutation fluctuated between isolates. In month 25 of treatment (TB6), we registered a decrease in the frequency of clones with gyrA D94H mutation and the rise of a new population of clones carrying gyrA A90V mutation (Fig. 3). Taking advantage of the closeness of the 2 variant positions, we examined whether the same clonal variant harboured these 2 SNP simultaneously. We did not find evidence of a single clonal variant harbouring these 2 variants together in any of the 97 reads that shared both positions. Clones carrying gyrA A90V vanished in subsequent isolates along with the implementation of adequate therapy. Although we detected both mutations 15 months apart, these resistance conferring-mutations emerged independently from the parental MDR strain (Fig. 1b and Fig. 3).

Frequency of drug-resistance conferring mutations along with treatment history. TERI: terizidone, PAS: para-aminosalicylic acid, OFX: ofloxacin, KAN: kanamycin, PZA: pyrazinamide, Cs: cycloserine, LNZ: linezolid, MXF: moxifloxacin, MXF-hd: moxifloxacin high dose, IC: imipenem-clavulanate, MC: meropenem-clavulanate AMK: amikacin. Continue-line: fixed mutations, dashed lines: non fixed mutations
When the therapy was resumed (52 months of treatment, TB7), we observed an increased genetic heterogeneity, involving both, short-term and fixed drug-resistance conferring mutations. Among the short-term SNPs, we found 2 mutations related to drug resistance, ethA (T88fs) and Rv0678 (N47fs). Mutations in the ethA gene have been reported to confer resistance to ETH [33]. More than half of the reads of TB7 and TB8 harboured an ethA frameshift mutation but these last 2 isolates remained notably susceptible to ETH. The patient’s drug scheme included ETH, but she took it irregularly due to intolerance until it was suspended. The Rv0678 gene – where we found a frameshift – is a transcriptional repressor of the genes encoding the MmpS5-MmpL5 efflux pump, which has been associated with bedaquiline (BDQ) and clofazimine (CFZ) resistance.
Among the drug resistance-conferring mutations that became fixed, one appeared towards the end of the treatment at the pncA gene (T177fs), confirming the phenotypic resistance to PZA. Another was gyrA D94H, which finally re-emerged and became fixed in the bacillary population (TB8) (Fig. 3). A mutation in the 5′ untranslated region of whiB7 gene (T-54G) was found in the last 2 isolates (TB7 and TB8). The whiB7 gene is a transcriptional activator of the eis gene. Mutations in its 5′ untranslated region have been related to whiB7 overexpression and the subsequent increase in eis expression, ultimately conferring resistance to KAN [34].
Within-host diversity/microevolution
To explore the within-host diversity among the 8 time-serial isolates, we called variants at a frequency of 25%. Along with the aforementioned short-term and fixed mutations related to drug resistance phenotypes, we found other 12 genomic variants: 6 changes were miss-sense mutations, 3 were InDels and 3 were synonymous mutations. (Supplementary Table 1). The overall substitution rate (5.68 SNPs per genome per year) was higher than the estimate for M. tuberculosis (0.4–0.5 SNPs per genome per year) [35]. This is in line with previous reports suggesting that antibiotics can distort the mutation rate, as random SNPs emerging in the genetic background of resistant clones could potentially fix together with well-established resistance-conferring mutations during the course of the treatment [4].
Looking at the identity of relevant variants, we found miss-sense mutations and InDels related to intermediary metabolism and respiration (oplA V674fs, Rv2141c T178A), a stress protein (Rv1636 D15A), lipid metabolism (pks6 G807C), and information pathways (recC A773D) (Supplementary Table 1), reinforcing the idea of within-host coexistence of different bacillary sub-populations of a single strain.
Discussion and conclusions
We studied 8 time-serial M. tuberculosis isolates from a patient with a previous TB history over 57 months of irregular treatment. According to the spoligotyping classification, all isolates belonged to the international shared type (SIT) 450, which has different geographic representation in the Americas, Europe, and Africa (http://www.pasteur-guadeloupe.fr:8081/SITVIT2/servletTools, last acceded 23rd March 2020). In Argentina, SIT 450 is present in low frequency in drug-susceptible and resistant M. tuberculosis clinical isolates [36]. Complete conservation of MIRU-VNTR and spoligotyping pattern was observed trough all samples. In line with the well-documented clonal stability of these markers, is the most likely situation among serial isolates from a single infection [37]. Moreover, the patient was born in Argentina, a country out of the list of high burden countries and had no known risk of TB exposure. These features make unexpected a mixed or superinfection scenario, typically seen in high incidence populations or high-risk settings [38]. In order to confirm that all isolates form a single monophyletic clade, we constructed a maximum-likelihood core genome phylogenetic tree. According to a recent SNP barcode-based phylogeny, all 8 isolates belong to L4 − the Euro-American lineage amply predominant in Latin America − and compose a monophyletic cluster within the barcode 4.1.1, which corresponds to the X type [31]. Taken together, these data strongly support intrapatient microevolution in a clonal population from a single infection.
In our patient, the microevolution process was mainly driven by short-term mutations which emerged and vanished along with variations in treatment efficacy and compliance. The most recent common ancestor was already MDR and evolved to pre-XDR over 7 months due to an inadequate treatment scheme.
Fluoroquinolone resistance, backed by the gyrA D94H substitution, was the earliest second line resistance to emerge, probably as a result of therapy including OFX and the consequent selection of resistant clones within the bacillary population. The gyrA D94H mutation confers high levels of resistance to LVX and MXF. In turn, the gyrA A90V mutation causes full resistance to LVX and low-levels or no resistance to MXF, which is still useful at high doses. In our case, even at the point when a borderline phenotypic resistance to MXF was detected, MXF was not excluded from the therapy scheme. This could be a reason why the clones harboring D94H mutation were selected within the bacillary population over the following months. The presence of 2 different clones harboring different fluoroquinolones resistance conferring-mutations supports the hypothesis of within-host independent microevolution events in the bacillary population. This is expected to occur in long-term therapy such as the case described here, in which the different drugs schemes successively administered, and the irregular treatment adherence shaped the evolution of bacilli subpopulations. This applies particularly to chronic recurrent pulmonary TB, which often involves multifocal lesions with different degrees of evolution and drug penetration. Within a single host, M. tuberculosis might benefit from this dynamic spatial and temporal heterogeneity by allowing the emergence of alternative drug-resistant variants according to the selective pressure exerted at different times in the different lesional microenvironments [39].
Allegedly, mutations in Rv0678 confer cross-resistance to BDQ and CFZ [40]. We detected a variant with a frameshift (N47fs) in this gene, but those drugs had never been administered to our patient. BDQ has been barely used in Argentina and was not available to the National TB Program at the time of the study. For this reason, we could not test the isolates for phenotypic susceptibility to this drug. On the other hand, CFZ is being included in M/XDR-TB drug schemes. We performed DST for CFZ and found the isolates carrying the mutation to be phenotypically resistant to CFZ (Supplementary Table 2). Variants in the Rv0678 gene were already described in BDQ and CFZ naïve patients in other settings [41, 42]. An explanation for these findings could be the emergence of random mutations in the within-host bacillary populations that were selected with the antibiotic therapy administered. This view is supported by the work of Villelas et al. [41] who found that Rv0678 variants associated with CFZ and BDQ resistance were more frequent in MDR isolates than in drug susceptible isolates from CFZ and BDQ naïve TB patients.
To our knowledge, the variant T-54G found herein in whiB7, had not been previously described, but a variant in the immediate previous position, G-55A, was reported to confer resistance to KAN [34]. TB8, one of the isolates carrying the novel whiB7 variant, was the subject of a noticeable discrepancy between results of gold standard DST methodologies and a phage-based technique [43]. This isolate resulted susceptible to KAN when tested by the proportion method and MIC (0.5 mg/L) at the TB NRL but was classified as resistant by a novel fluoro-mycobacteriophage reporter method when performed at Hospital Muñiz. We speculate that mutation T-54G in whiB7 might be associated with low-level resistance to KAN missed by standard growth-based methods, as described for certain non-canonical rpoB mutations in relation to phenotypic RIF resistance [44].
Another interesting variant was G807C in pks6 found in TB6. The pks gene family codifies for diverse polyketide synthases. This gene family was shown to have a wide within-host variation [43]. It was proposed that the unusual variability of these genes could allow M. tuberculosis to manipulate the host cell during infection. In turn, it is disease severity what seems to drive within-host diversity of pks genes rather than M. tuberculosis lineage, TB treatment, or drug resistance status [45].
It is common knowledge that incomplete, intermittent, or inadequate treatment promotes the development of MDR- and XDR-TB. Notably, some recent reports from high-income countries where TB has low incidence have described microevolution leading to drug resistance even in single patients with treatment adherence [4, 46]. In medium and low-income countries, the situation is more propitious to drug resistance. Even where anti-TB drugs are available, in most developing countries TB treatment programs are deficient, usually because of poor treatment compliance of patients and failure to carefully sustain drug supplies and observed treatment. The genomic scrutiny of isolates from TB patients in this context can contribute to a better understanding of the evolution mechanisms of M. tuberculosis strains leading to drug resistance. Based on this work and previous data, we emphasize and recommend adequate treatment as preventing the emergence of MDR and XDR TB.
Availability of data and materials
The dataset supporting the conclusions of this article is available in the SRA NCBI repository [Accession number PRJNA646920, https://www.ncbi.nlm.nih.gov/bioproject/PRJNA646920].
Abbreviations
- AFB:
-
Acid-fast bacilli
- AMGs :
-
Aminoglycosides.
- AMK:
-
Amikacin
- ANLIS:
-
National institute of infectious diseases Dr. Carlos Malbrán
- CFZ:
-
Clofazimine
- Cs :
-
Cycloserine
- DST:
-
Drug susceptibility testing
- EMB:
-
Ethambutol
- ETH :
-
Ethionamide
- IC:
-
Imipenem-clavulanate
- InDels:
-
Insertion–deletion mutations
- INH:
-
Isoniazid
- KAN:
-
Kanamycin
- LNZ :
-
Linezolid
- LVX:
-
Levofloxacin
- MC: :
-
Meropenem-clavulanate
- MDR-TB:
-
Multidrug-resistant tuberculosis
- MIRU:
-
Mycobacterial interspersed repetitive units
- MXF:
-
Moxifloxacin
- NRL:
-
National reference laboratory
- OFX :
-
Ofloxacin
- PAS :
-
Para-aminosalicylic acid
- PZA:
-
Pyrazinamide
- RIF:
-
Rifampicin
- SIT:
-
International shared type
- SNP:
-
Single nucleotide polymorphism
- STR:
-
Streptomycin
- TB:
-
Tuberculosis
- TERI :
-
Terizidone
- VNTR:
-
Variable number tandem repeat
- XDR-TB:
-
Extensively drug-resistant tuberculosis
References
Horsburgh CR, Mitnick CD, Lange C. The MDR-TB epidemic—a status report. Int J Tuberc Lung Dis. 2019;23:121–2. https://doi.org/10.5588/ijtld.18.0000.
Trauner A, Liu Q, Via LE, Liu X, Ruan X, Liang L, et al. The within-host population dynamics of Mycobacterium tuberculosis vary with treatment efficacy. Genome Biol. 2017;18. https://doi.org/10.1186/s13059-017-1196-0.
Dookie N, Rambaran S, Padayatchi N, Mahomed S, Naidoo K. Evolution of drug resistance in Mycobacterium tuberculosis: a review on the molecular determinants of resistance and implications for personalized care. J Antimicrob Chemother. 2018;73:1138–51.
Eldholm V, Norheim G, von der Lippe B, Kinander W, Dahle UR, Caugant DA, et al. Evolution of extensively drug-resistant Mycobacterium tuberculosisfrom a susceptible ancestor in a single patient. Genome Biol. 2014;15. https://doi.org/10.1186/s13059-014-0490-3.
Cohen KA, Manson AL, Abeel T, Desjardins CA, Chapman SB, Hoffner S, et al. Extensive global movement of multidrug-resistant M. tuberculosis strains revealed by whole-genome analysis. Thorax. 2019;74:882–9. https://doi.org/10.1136/thoraxjnl-2018-211616.
Lipworth S, Hough N, Leach L, Morgan M, Jeffery K, Andersson M, et al. Whole-genome sequencing for predicting clarithromycin resistance in Mycobacterium abscessus. Antimicrob Agents Chemother. 2019. https://doi.org/10.1128/AAC.01204-18.
Eldholm V, Monteserin J, Rieux A, Lopez B, Sobkowiak B, Ritacco V, et al. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat Commun. 2015;6:7119.
Cohen KA, Abeel T, Manson McGuire A, Desjardins CA, Munsamy V, Shea TP, et al. Evolution of extensively drug-resistant tuberculosis over four decades: whole genome sequencing and dating analysis of mycobacterium tuberculosis isolates from KwaZulu-Natal. PLoS Med. 2015;12:e1001880.
Gardy JL, Johnston JC, Ho Sui SJ, Cook VJ, Shah L, Brodkin E, et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011;364:730–9.
Muwonge A, Kankya C, Olea-Popelka F, Biffa D, Ssengooba W, Berit D, et al. Molecular investigation of multiple strain infections in patients with tuberculosis in Mubende district, Uganda. Infect Genet Evol. 2013;17:16–22.
Ritacco V, Di Lonardo M, Reniero A, Ambroggi M, Barrera L, Dambrosi A, et al. Nosocomial Spread of Human Immunodeficiency Virus‐Related Multidrug‐Resistant Tuberculosis in Buenos Aires. J Infect Dis. 1997;176:637–42. https://doi.org/10.1086/514084.
Monteserin J, Pérez-Lago L, Yokobori N, Paul R, Maus SR, Simboli N, et al. Trends of Two Epidemic Multidrug-Resistant Strains of Mycobacterium tuberculosis in Argentina Disclosed by Tailored Molecular Strategy. Am J Trop Med Hyg. 2019;101:1308–11. https://doi.org/10.4269/ajtmh.19-0397.
Organization WH, Others. Technical report on critical concentrations for drug susceptibility testing of medicines used in the treatment of drug-resistant tuberculosis. Geneva: World Health Organization; 2018. p. 21 https://apps.who.int/iris/bitstream/handle/10665/260470/WHO-CDS-TB-2018.5-eng.pdf.
Paul R, Masciotra N, Lafuente G, Avendaño D, Monteserin J, Belen OM, et al. Field validation of multiplex allele-specific PCR (MAS-PCR) for rapid detection of multidrug-resistant tuberculosis (MDR_TB) in Argentina. F1000Res. 2015;6. https://f1000research.com/posters/1097888.
Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 1997;35:907–14.
Supply P, Allix C, Lesjean S, Cardoso-Oelemann M, Rüsch-Gerdes S, Willery E, et al. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol. 2006;44:4498–510.
Demay C, Liens B, Burguière T, Hill V, Couvin D, Millet J, et al. SITVITWEB – A publicly available international multimarker database for studying Mycobacterium tuberculosis genetic diversity and molecular epidemiology. Infect Genet Evol. 2012;12:755–66. https://doi.org/10.1016/j.meegid.2012.02.004.
Weniger T, Krawczyk J, Supply P, Niemann S, Harmsen D. MIRU-VNTRplus: a web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 2010;38(Web Server issue):W326–31.
van Soolingen D, Hermans PW, de Haas PE, Soll DR, van Embden JD. Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J Clin Microbiol. 1991;29:2578–86.
Wingett SW, Andrews S. FastQ screen: a tool for multi-genome mapping and quality control. F1000Res. 2018;7:1338.
Cantu VA, Sadural J, Edwards R. PRINSEQ, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. https://doi.org/10.7287/peerj.preprints.27553.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92.
Coll F, McNerney R, Preston MD, Guerra-Assunção JA, Warry A, Hill-Cawthorne G, et al. Rapid determination of anti-tuberculosis drug resistance from whole-genome sequences. Genome Med. 2015;7:51.
Martí-Carreras J, Maes P. VANIR—NextFlow Pipeline for Viral Variant Calling and de Novo Assembly of Nanopore and Illumina Reads for High-Quality dsDNA Viral Genomes. Proceedings. 2020;50:117. https://doi.org/10.3390/proceedings2020050117.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45:D566–73.
Sosa EJ, Burguener G, Lanzarotti E, Defelipe L, Radusky L, Pardo AM, et al. Target-Pathogen: a structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic Acids Res. 2018;46:D413–8.
Sekizuka T, Yamashita A, Murase Y, Iwamoto T, Mitarai S, Kato S, et al. TGS-TB: total 22 genotyping solution for mycobacterium tuberculosis using short-read whole-genome sequencing. PLoS One. 2015;10:e0142951.
Stamatakis A. Using RAxML to Infer phylogenies. Curr Prot Bioinform. 2015:6–14. https://doi.org/10.1002/0471250953.bi0614s51 6.14.1.
Coll F, McNerney R, Guerra-Assunção JA, Glynn JR, Perdigão J, Viveiros M, et al. A robust SNP barcode for typing Mycobacterium tuberculosis complex strains. Nat Commun. 2014;5:4812.
Brynildsrud OB, Pepperell CS, Suffys P, Grandjean L, Monteserin J, Debech N, et al. Global expansion of lineage 4 shaped by colonial migration and local adaptation. Sci Adv. 2018;4:eaat5869.
Vilchèze C, Jacobs WR Jr. Resistance to isoniazid and ethionamide in mycobacterium tuberculosis: genes, mutations, and causalities. Microbiol Spectr. 2014;2 MGM2–0014 – 2013;2(4):1–21.
Reeves AZ, Campbell PJ, Sultana R, Malik S, Murray M, Plikaytis BB, et al. Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5’ untranslated region of whiB7. Antimicrob Agents Chemother. 2013;57:1857–65.
Eldholm V, Balloux F. Antimicrobial Resistance in Mycobacterium tuberculosis : The Odd One Out. Trends Microbiol. 2016;24:637–48. https://doi.org/10.1016/j.tim.2016.03.007.
Monteserin J, Paul R, Gravina E, Reniero A, Hernandez T, Mazzeo E, et al. Genotypic diversity of Mycobacterium tuberculosis in Buenos Aires, Argentina. Infect Genet Evol. 2018;62:1–7. https://doi.org/10.1016/j.meegid.2018.04.006.
Al-Hajoj SAM, Akkerman O, Parwati I, Al-Gamdi S, Rahim Z, van Soolingen D, et al. Microevolution of Mycobacterium tuberculosis in a tuberculosis patient. J Clin Microbiol. 2010;48:3813–6.
Shamputa IC, Jugheli L, Sadradze N, Willery E, Portaels F, Supply P, et al. Mixed infection and clonal representativeness of a single sputum sample in tuberculosis patients from a penitentiary hospital in Georgia. Respir Res. 2006;7:99.
Lenaerts A, Barry CE 3rd, Dartois V. Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol Rev. 2015;264:288–307.
Ismail N, Omar SV, Ismail NA, Peters RPH. Collated data of mutation frequencies and associated genetic variants of bedaquiline, clofazimine and linezolid resistance in Mycobacterium tuberculosis. Data Brief. 2018;20:1975–83. https://doi.org/10.1016/j.dib.2018.09.057.
Villellas C, Coeck N, Meehan CJ, Lounis N, de Jong B, Rigouts L, et al. Unexpected high prevalence of resistance-associated Rv0678 variants in MDR-TB patients without documented prior use of clofazimine or bedaquiline. J Antimicrob Chemother. 2017;72:684–90.
Klopper M, Heupink TH, Hill-Cawthorne G, Streicher EM, Dippenaar A, de Vos M, et al. A landscape of genomic alterations at the root of a near-untreatable tuberculosis epidemic. BMC Med. 2020;18:24.
Rondón L, Urdániz E, Latini C, Payaslian F, Matteo M, Sosa EJ, et al. Fluoromycobacteriophages Can Detect Viable Mycobacterium tuberculosis and Determine Phenotypic Rifampicin Resistance in 3–5 Days From Sputum Collection. Front Microbiol. 2018;9. https://doi.org/10.3389/fmicb.2018.01471.
Van Deun A, Barrera L, Bastian I, Fattorini L, Hoffmann H, Kam KM, et al. Mycobacterium tuberculosis strains with highly discordant rifampin susceptibility test results. J Clin Microbiol. 2009;47:3501–6.
O’Neill MB, Mortimer TD, Pepperell CS. Diversity of Mycobacterium tuberculosis across Evolutionary Scales. PLoS Pathog. 2015;11:e1005257.
Liu Q, Via LE, Luo T, Liang L, Liu X, Wu S, et al. Within patient microevolution of Mycobacterium tuberculosis correlates with heterogeneous responses to treatment. Sci Rep. 2015;5:17507.
Acknowledgments
We are grateful to Peter Draschman for English correction.
Funding
Agencia Nacional de Promoción Científica y Tecnológica [ANPCyT, PICT-2018-04663 to D.F.D.P.], Centro Latinoamericano de Formación Interdisciplinaria 2017 ´[to D.F.D]. CONICET membership of the research career [D.F. D;.J. M.; E. J. S.; N.Y.; M. M.; B. L.; A. T. and V. R.], CONICET doctoral fellowship [A.P. and F.S]. The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
D.A.F.D.P, J.M., J.C., E. S., M. M, A.T.,B. L., and V.R. conceived the study design. J.M., J.C., M.M., N.Y., T.P., I.W., N.S., R.P, B.L. and V.R. performed wet lab experimental work. D.A.F.D.P., J.M., E.S., M.M., F.S., A.F.B., A.P., M.M and A.T., performed bioinformatic analysis. D.A.F.D.P., J.M., A.G.T. and V.R. drafted the manuscript with input from the other authors. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This research has been approved by the INEI ANLIS research review board.
Consent for publication
The patient provided written consent for the publication of the data including her clinical details.
Competing interests
The authors declare that they have no competing interests
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1
. Within-host microevolution of eight time-serial isolates from a patient with multidrug-resistant tuberculosis and inadequate compliance to treatment
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fernandez Do Porto, D.A., Monteserin, J., Campos, J. et al. Five-year microevolution of a multidrug-resistant Mycobacterium tuberculosis strain within a patient with inadequate compliance to treatment. BMC Infect Dis 21, 394 (2021). https://doi.org/10.1186/s12879-021-06069-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-021-06069-9