
Abstract
Background
Quinine (QN) remains the first line anti-malarial drug for the treatment of complicated malaria in Europe and Africa. The emergence of QN resistance has been documented. QN resistance is not yet a significant problem, but there is an urgent need to discover partners for use in combination with QN. The aim of the study was to assess the in vitro potentiating effects of atorvastatin (AVA), a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, in combination with QN against Plasmodium falciparum and to evaluate whether the effects of AVA could be associated with gene copy number or mutations in genes involved in QN resistance, such as pfcrt, pfmdr1, pfmrp and pfnhe.
Methods
The susceptibilities to combination of AVA with QN were assessed against 21 parasite strains using the in vitro isotopic microtest. Genotypes and gene copy number were assessed for pfcrt, pfmdr1, pfmdr2, pfmrp genes. In addition, the number of DNNND, DDNHNDNHNN repeats in pfnhe-1 ms4760 and the ms4760 profile were determined for each strains of P. falciparum.
Results
AVA demonstrated synergistic effects in combination with QN against 21 P. falciparum strains. The QN IC50 was reduced by 5% (0% to 15%; 95%CI: 1%-8%), 10% (3% to 23%; 95%CI: 7%-14%) and 22% (14% to 40%; 95%CI: 19%-25%) in presence of AVA at concentrations of 0.1, 0.5 and 1.0 μM, respectively. These reductions were all significant (p < 0.009). The reduction in the QN IC50 in presence of AVA was not significantly correlated with the QN IC50 (r = 0.22, P = 0.3288) or the AVA IC50 (r = 0.03, P = 0.8946). The synergistic effect of AVA in combination with QN was not significantly associated with polymorphisms in the pfcrt, pfmdr1, pfmrp, and pfnhe-1 genes that could be involved in QN resistance. The synergistic effect of AVA on QN responses was not significantly associated with pfmdr1 copy number (P = 0.0428).
Conclusion
The synergistic effect of AVA in combination with QN was found to be unrelated to mutations occurring in transport protein genes involved in QN drug resistance. The different mechanisms of drug uptake and/or mode of action for AVA compared to the other anti-malarial drugs, as well as the AVA-mediated synergy of the anti-malarial effect of QN, suggests that AVA will be a good candidate for combinatorial malaria treatment. All of these observations support calls for both an in vivo evaluation with pharmacokinetic component and clinical trials of AVA as an anti-malarial therapy.
Similar content being viewed by others

Background
During the past 20 years, many strains of Plasmodium falciparum have become resistant to chloroquine and other anti-malarial drugs [1]. This has prompted a search for an effective alternative anti-malarial drug with minimal side effects. The emergence and spread of parasites resistant to anti-malarial drugs has resulted in an urgent need for discovery and development of novel anti-malarial compounds.
Quinine (QN) has been used as a malaria treatment for more than 350 years in Africa and remains the first-line anti-malarial drug for the treatment of complicated malaria in Europe and Africa. However, cases of QN clinical failure were observed in Brazil and Asia in the 1960s [2, 3]. In the 1980s, clinical failures became more frequent in Southeast Asia, South America and Africa [4–8]. However, QN resistance is not yet a significant problem. QN continues to be widely used at present as a second-line therapy for uncomplicated malaria in Africa and other areas. Nevertheless, there is an urgent need for discovery partners for combination with QN.
Atorvastatin (AVA), a 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase inhibitor, belongs to a family of lipid-lowering drugs that are currently used for the control of hyperlipidaemia and are considered useful for protection from cardiovascular events. Apart from the cholesterol-lowering activity of statins, the immunomodulatory and pleiotropic effects of statins may significantly impact infection-related survival [9, 10]. AVA reduced the intracellular growth of Salmonella typhimurium in macrophages in a mouse model [11]. Additionally, AVA has an anti-hepatitis C virus (HCV) effect and enhanced the anti-HCV effect of interferon when used in combination [12]. However, the use of statin alone in patients with, or at risk of, severe infections is contentious [13]. Preoperative statin use was not associated with a reduction of the rate of postoperative infection among patients who underwent cardiac surgery.
Statins severely interfered with the growth of protozoan parasites in the Trypanosomatidae family, such as Trypanosoma cruzi and various Leishmania species [14–16]. Additionally, an HMG-CoA reductase has been detected in Trypanosoma and Leishmania[16].
AVA demonstrated anti-malarial activity in vitro even though an HMG-CoA homolog was not identified by BLASTX analysis of the P. falciparum sequence with other protozoal HMG-CoA protein sequences [17]. AVA is ten-fold more active against P. falciparum strains than other statins [17]. Additionally, AVA demonstrated no in vitro cross-resistance with quinine, chloroquine, monodesethylamodiaquine, mefloquine, lumefantrine, dihydroartemisinin, atovaquone, or doxycycline, and the IC50 values for AVA were found to be unrelated to mutations in transport protein genes involved in quinoline anti-malarial drug resistance, such as pfcrt, pfmdr1, pfmrp, and pfnhe-1[18]. AVA had synergistic effects in combination with dihydroartemisinin [19].
AVA is also an inhibitor of phosphoglycoprotein (Pgp), an efflux protein in cancer cells [20–22]. Multi-drug resistance (MDR)-like proteins are involved in quinoline resistance in P. falciparum[23–25]. PfMRP is involved in QN resistance [26, 27].
The objectives of this study were 1) to assess the in vitro potentiation effects of AVA in combination with QN against 21 strains of P. falciparum that were isolated from a wide panel of countries and had different drug susceptibility profiles, and 2) to evaluate if AVA could be an inhibitor of P. falciparum MDR-like proteins, such as Pgh1, PfMRP or PfMDR2, or transporters involved in drug resistance, such as PfCRT (P. falciparum chloroquine resistance transporter) [28] and PfNHE-1 (P. falciparum sodium/hydrogen exchanger) [29, 30].
Methods
Plasmodium falciparum cultures
Twenty-one parasite strains or clones from a wide panel of countries (Brazil, Cambodia, Cameroon, Djibouti, French Guyana, the Gambia, Honduras, Indochina, Niger, Republic of Comoros, Republic of the Congo, Senegal, Sierra Leone, Sudan, and Uganda) were maintained in culture in RPMI 1640 (Invitrogen, Paisley, UK) supplemented with 10% human serum (Abcys S.A. Paris, France) and buffered with 25 mM HEPES and 25 mM NaHCO3. Parasites were grown in type A+ human red blood cells under controlled atmospheric conditions (10% O2, 5% CO2, and 85% N2) at 37°C with a humidity of 95%. All strains were synchronized twice with sorbitol before use [31]. Clonality was verified using PCR genotyping of polymorphic genetic markers, msp1, msp2, and microsatellite loci [32, 33]. The potentiation evaluation for each strain was assessed in three independent experiments.
Drugs
AVA calcium salt was purchased from Molekula (UK). QN was purchased from Sigma (Saint Louis, MO). AVA was dissolved in 1% DMSO (v/v) in RPMI. Two-fold serial dilutions, with final concentrations ranging from 0.01 μM to 200 μM, were prepared in 1% DMSO in RPMI and plated in 96-well plates just before use. QN was dissolved first in methanol and then diluted in water to obtain final concentrations ranging from 5 to 3400 nM.
In vitro assay
In order to assess the synergy of AVA with QN, 25 μl of AVA per well and 200 μl of the suspension of synchronous parasitized red blood cells (final parasitaemia, 0.5%; final haematocrit, 1.5%) per well were plated in 96-well plates containing the QN concentrations. The in vitro isotopic micro-test used had been previously described [34].
The drug concentration able to inhibit 50% of parasite growth (IC50) was calculated as the drug concentration corresponding to 50% of the incorporation of tritiated hypoxanthine by the parasite in the drug-free control wells. The IC50 value was determined using the non-linear regression analysis of log-based dose-response curves (Riasmart, Packard, Meriden, USA).
In order to evaluate the modulation of QN resistance by AVA, isobolograms were constructed by plotting a pair of fractional IC50 for each combination of AVA and QN for both parasite strains. The AVA fractional IC50 was calculated by dividing their fixed concentrations by the IC50 of tested drugs alone and plotted on the horizontal axis. The QN fractional IC50 was calculated by dividing the IC50 of QN combined with fixed concentrations of AVA and plotted on the vertical axis. Points lying above the diagonal line (corresponding to the points where there is no interaction between the drugs) are antagonistic, points below the diagonal line are considered to be synergistic.
The interaction parameter indicating relative degree of interaction (I) was calculated using the equation Yi = 1-[Xi/(Xi + ei* (1 - Xi))], where Yi = IC50 for AVA when combined with QN and Xi = IC50 for QN when combined with AVA [35]. This representation of drug interaction provides a single quantitative estimate of relative degree of interaction even when the experimental ratios are not optimum. Positive values of I indicate synergism, negative values antagonism; addition occurs when it equals zero.
In addition, three concentrations of AVA (0.1, 0.5 and 1 μM), which were relevant with AVA plasma concentrations achievable in patients taking 80 mg of AVA daily [36], were reanalysed separately. The concentration of AVA that reduced the IC50 of QN by 50% when used alone, [AVA]QN, was calculated for each strain of P. falciparum.
Nucleic acid extraction
The total genomic DNA of each strain was isolated using the E.Z.N.A. Blood DNA kit (Omega Bio-Tek, GA, U.S.A.). The RNA of each strain was purified using the QIAamp Blood Mini kit (QIAGEN, Germany). The methods for the SNPs identification of pfcrt[37], pfmdr1[38], pfmdr2[38] and pfmrp[38], for the identification of pfnhe-1 microsatellite profiles [29, 37] and for the estimation of the copy number of pfmdr1 and pfmdr2[38] were previously described.
Statistical analysis
The differences in MQ IC50 between AVA concentrations groups have first been tested using ANOVA for repeated measures to take into account the fact that observations made within each strain were not independent. Using the most conservative correction for interdependence between observations (i.e. Box's conservative epsilon), the differences in QN IC50 were tested for concentrations relevant with AVA plasma concentrations achievable in patients taking 80 mg of AVA daily (0.1, 0.5 and 1 μM). Using a random effect linear regression approach, the regression coefficients for the log-transformed QN IC50% indicated the significance of the mean fold change in QN IC50 when adding AVA concentrations of 0.1, 0.5 and 1.0 μM.
The Kruskal-Wallis test or the Mann-Whitney U test of was used, when appropriate, to compare the equality of the populations for each mutation. The results of these tests were compared according to the alleles at each locus. The differences in IC50 for QN were then tested 22 times (i.e., one per locus). The probability of getting a significant result with 22 tests at the α = 0.05 level of significance was 1-0.9522 (1-probability of not getting a significant result with 22 tests). According to the Bonferroni correction, it was concluded that a difference was significant when at least one of the 22 comparisons yielded a significance level below 0.05/22 = 0.0023.
Results
Results of in vitro potentiation
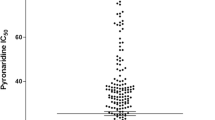
AVA demonstrated synergistic effects in combination with QN (Figure 1). The differences in QN IC50 between AVA concentrations groups have first been tested using ANOVA for repeated measures to take into account the fact that observations made within each strain were not independent. Using the most conservative correction for interdependence between observations (i.e. Box's conservative epsilon), the differences in QN IC50 were highly significant (p < 0.009) at concentrations relevant with AVA plasma concentrations achievable in patients taking 80 mg of AVA daily (Table 1). Using a random effect linear regression approach, the regression coefficients for the log-transformed QN IC50% indicated that the mean fold change in QN IC50 when adding AVA concentrations of 0.1, 0.5 and 1.0 μM (0.95; 0.90 and 0.78, respectively) were also highly significant (p < 0.009). The QN IC50 was reduced by 5% (0% to 15%; 95%CI: 1%-8%), 10% (3% to 23%; 95%CI: 7%-14%) and 22% (14% to 40%; 95%CI: 19%-25%) in presence of AVA at concentrations of 0.1, 0.5 and 1.0 μM, respectively. These reductions were all significant (p < 0.009). Another finding was that the synergy of the effects of QN was AVA dose-dependent. The reductions in QN IC50 were also significant between 0.1 AVA and 0.5 AVA (p < 0.0014) and between 0.5 AVA and 1.0 AVA (p < 0.0001).

In vitro synergistic interactions between atorvastatin (AVA) and quinine (QN) against the 21 strains of P. falciparum.
Eleven strains (53%) had interaction parameter indicating relative degree of interaction (I) > 1 (synergism) and 7 (33%) with 0 < I < 1 (weak synergism). Three strains (14%) had I ranging from -1 to 0 (weak antagonism).
The concentrations of AVA that reduced the IC50 of QN when used alone by 50% ([AVA]QN) ranged from 1.1 to 13.0 μM (geometric mean = 3.7 μM and 95% confidence interval = 2.8-5.1). The reduction of the QN IC50 in presence of AVA was not significantly correlated with the QN IC50 (r = 0.22, P = 0.3288) or the AVA IC50 (r = 0.03, P = 0.8946).
Genotypes and gene copy number of the 21 strains of P. falciparum
The following mutations were identified in at least one strain: pfcrt M74I, N75E, K76T, A220S, Q271(E/V), N326S, I356T, and I371R; pfmrp H191Y and S437A; pfmdr1 N86Y, Y184F, S1034C, N1042D, and D1246Y; and pfmdr2 S208N and F423Y. Eight different ms4760 microsatellite profiles of pfnhe-1 were observed. The number of DNNND and DDNHNDNHNN repeats in ms4760 ranged from one to four and one to two, respectively. The copy number of pfmdr1 ranged from one to three. Only one copy of pfmdr2 was found in all of the 21 P. falciparum strains.
Association of in vitro responses (IC50) and polymorphisms of genes
The association between polymorphism in pfcrt gene (codons 75, 76 and 220, P < 0.0022) or in pfmrp gene (codons 191 and 437, P = 0.0008) and QN responses, used alone, was highly significant (Additional file 1). The associations between ms4760 profiles, number of DNNND repeats or polymorphism in pfmdr1 gene (codons 1034 and 1042) and QN responses were not significant (P < 0.05 but > 0.0023) (Additional file 1). The synergistic effect of AVA on QN responses was not significantly associated with polymorphisms in the pfcrt, pfmdr1, pfmrp and pfnhe-1 genes (Additional file 1).
Association of in vitro responses (IC50) and gene copy number
The association between pfmdr1 copy number and QN responses was not significant (P = 0.0086) (Additional file 1). The synergistic effect of AVA on QN responses was not significantly associated with pfmdr1 copy number (P = 0.0428) (Additional file 1).
Discussion
Atorvastatin (AVA), a 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor, inhibited the in vitro growth of P. falciparum at the micromolar range. Whereas the generally agreed upon level of efficacy is in the low to mid-nanomolar range, if the mechanism of action of a novel anti-malarial compound was sufficiently new and different than those of the commonly used anti-malarial drugs, this compound would warrant further study. Nevertheless, AVA, used alone at 20 mg/kg of body weight, failed to prevent death from cerebral malaria or to effect parasitaemia in infected mice [39, 40].
A large amount of scientific effort is spent on elucidating the mechanisms underlying the resistance to anti-malarial drugs with the hope of restoring/improving the efficacy of existing drugs and developing new drugs that can bypass the resistance mechanisms. One of the strategies that has been used to reduce the prevalence of malaria is the use of drug combinations. The combination protects each drug from the development of resistance and reduced the overall transmission rate of malaria [41]. The absence of in vitro cross-resistance between AVA and QN or the other anti-malarial drugs suggested that AVA could be a good potential partner for these anti-malarial drugs [18]. These different considerations and the capacity of AVA to inhibit human Pgh [22] lead us to evaluate AVA in combination with QN. AVA potentiated the activity of QN against 86% of the P. falciparum parasites tested (53% of synergism and 33% of weak synergism). However, AVA did not improve the QN responses of the totality of the strains tested. The potentiation was independent of the QN susceptibility level. AVA improved the in vitro activity of QN at concentrations relevant with AVA plasma concentrations achievable in patients taking 80 mg of AVA daily [36]. Furthermore, the administration doses of AVA in humans could be increased to 120 mg daily with only limited additional side effects [42]. A dose of 120 mg of AVA increased the maximal plasma concentration of Cmax after oral administration by levels from 4 to 10 fold. Cerebral malaria shares common pathophysiological features with sepsis, especially with regard to the pathology of the endothelium [43], and critically ill patients with sepsis had a significantly higher Cmax when compared to the healthy volunteers (110.5 vs. 5.9 ng/ml) [44]. The most important adverse events related to the use of atorvastatin are muscle toxicity and the effects on liver enzymes. However, rhabdomyolysis and creatinine kinase or transaminase level were not significantly increased in trials of intensive statin therapy or at high dose of AVA (80 mg/day) for periods ranging from 2 weeks to 5 years and were not age related [45–48]. The administration doses of AVA in humans at 120 mg daily did not induce rhabdomyolysis [42]. Cases of rhabdomyolysis were observed when AVA was co-administrated with other drugs, such as fusidic acid or ezetimibe or in patients with nephrotic syndrome. Biochemical evidence of skeletal muscle damage was common in malaria, but rhabdomyolysis appeared to be rare [49–51]. Rhabdomyolysis should be carefully survey in co-administration of AVA in malaria.
Nevertheless, these effects were not associated with polymorphisms in the genes involved in QN resistance such as pfcrt[28, 52], pfmdr1[53], pfmrp[26, 54], or pfnhe-1[29, 30, 55]. The synergistic effect of AVA in combination with QN was found to be unrelated to mutations occurring in the transport protein genes involved in quinoline drug resistance. In addition, the synergistic effect of AVA on QN responses was not significantly associated with pfmdr1 copy number but, as QN resistance, the significance level of this association was above the Bonferroni-corrected P value threshold (0.0023) but below 0.05. These data suggested that AVA could interfere with Pgh1, as predicted by the inhibition of Pgh in human cells by AVA [20–22]. However, this is a preliminary report. Twenty-one strains may not be sufficient to make definite conclusions.
Synergy of QN with AVA is a promising result in malaria treatment. QN remains the first line anti-malarial drug for the treatment of complicated malaria in Europe and Africa. However, clinical failures became more frequent in Southeast Asia, South America and Africa [4, 6, 8]. Artemisinin-based combination therapy (ACT) was proposed as a first-line treatment for uncomplicated malaria six years ago. However, individual P. falciparum isolates resistant to artemisinin in vitro and the first clinical failures have been described in Cambodia [56–60]. This emergence of parasite resistance to ACT indicates that novel compounds and combinations need to be discovered and developed. These data suggest that AVA will be a good candidate for combination with QN in malaria treatment.
Conclusions
The mechanism underlying the anti-malarial activity of AVA is currently unknown. The absence of cross-resistance with QN, chloroquine, monodesethylamodiaquine, mefloquine, lumefantrine, dihydroartemisinin, atovaquone, or doxycycline suggests a different mechanism of drug uptake and/or mode of action for AVA [18]. AVA is a 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Nevertheless, an HMG-CoA homolog was not identified by BLASTX analysis of the P. falciparum genome with other protozoal HMG-CoA proteins sequences. Parasites treated with mevastatin demonstrated depressed biosynthesis of dolichol and isoprenoid pyrophosphate [61]. In addition, mevastatin decreased the viability of cells through inhibition of the proteasome. One of the future objectives is to identify modifications of the P. falciparum proteome in parasites treated with AVA.
The different mechanisms of drug uptake and/or mode of action for AVA compared to the other anti-malarial drugs, as well as the AVA-mediated potentiation of the anti-malarial effect of QN, suggests that AVA will be a good candidate for combinatorial malaria treatment. All of these observations support calls for both an in vivo evaluation with pharmacokinetic component and clinical trials of AVA as an anti-malarial therapy.
Conflict of interests
The authors have no conflicts of interest concerning the work reported in this paper. The authors do not own stocks or shares in a company that might be financially affected by the conclusions of this article.
References
Le Bras J, Musset L, Clain J: Antimalarial drug resistance. Med Maladies Infect. 2006, 36: 401-405. 10.1016/j.medmal.2006.05.005.
Bjorkman A, Phillips-Howard PA: The epidemiology of drug-resistant malaria. Trans R Soc Trop Med Hyg. 1990, 84: 177-180. 10.1016/0035-9203(90)90246-B.
Giboda M, Denis MB: Response of Kampuchean strains of Plasmodium falciparum to antimalarials: in-vivo assessment of quinine and quinine plus tetracycline; multiple drug resistance in vitro. J Trop Med Hyg. 1988, 91: 205-211.
Harinasuta T, Bunnag D, Lasserre R: Quinine resistant falciparum malaria treated with mefloquine. Southeast Asian J Trop Med Public Health. 1990, 21: 552-557.
Molinier S, Imbert P, Verrot D, Morillon M, Parzy D, Touze JE: Plasmodium falciparum malaria: type R1 quinine resistance in East Africa. Presse Med. 1994, 23: 1494-
Jelinek T, Schelbert P, Löscher T, Eichenlaub D: Quinine resistant falciparum acquired in east Africa. Trop Med Parasitol. 1995, 46: 38-40.
Tish KN, Pillans PI: Recrudescence of Plasmodium falciparum malaria contracted in Lombok, Indonesia after quinine/doxycycline and mefloquine: case report. N Z Med J. 1997, 110: 255-256.
Pradines B, Pistone T, Ezzedine K, Briolant S, Bertaux L, Receveur MC, Parzy D, Millet P, Rogier C, Malvy D: Qunine-resistant malaria in traveler returning from Senegal, 2007. Emerg Infect Dis. 2010, 16: 546-548. 10.3201/eid1603.091669.
Terblanche M, Almog Y, Rosenson RS, Smith TS, Hackam DG: Statins: panacea for sepsis?. Lancet Infect Dis. 2006, 6: 242-248. 10.1016/S1473-3099(06)70439-X.
Greenwood J, Mason JC: Statins and the vascular endothelial inflammatory response. Trends Immunol. 2007, 28: 88-98. 10.1016/j.it.2006.12.003.
Catron DM, Lange Y, Borenztajn J, Sylvester MD, Jones BD, Haldar K: Salmonella enterica serovar Typhimurium requires nonsterol precursors of the cholesterol biosynthetic pathway for intracellular proliferation. Infect Immun. 2004, 72: 1036-1042. 10.1128/IAI.72.2.1036-1042.2004.
Ikeda M, Kato N: Life style-related diseases of the digestive system: cell culture system for the screening of anti-Hepatitis C virus (HCV) reagents: suppression of HCV replication by statins and synergistic action with interferon. J Pharmacol Sci. 2007, 105: 145-150. 10.1254/jphs.FM0070050.
Mohamed R, McAlister FA, Pretorious V, Kapoor AS, Majumdar SR, Ross DB, Norris CM: Preoperative statin use and infection after cardiac surgery: a cohort study. Clin infect Dis. 2009, 48: 66-72. 10.1086/597300.
Urbina JA, Lazardi K, Marchan E, Visbal G, Aguirre T, Piras MM, Piras R, Maldonado RA, Payares G, de Souza W: Mevinolin (Lovastatin) potentiates the antiproliferative effects of ketoconazole and terbinafine against Trypanosoma (Schizotrypanum) cruzi: in vitro and in vivo studies. Antimicrob Agents Chemother. 1993, 37: 580-591.
Yokoyama K, Trobridge P, Buckner FS, Scholten J, Stuart KD, van Voorhis WC, Gelb MH: The effects of protein farnesyltransferase inhibitors on trypanosomatids: inhibition of protein farnesylation and cell growth. Mol Biochem Parasitol. 1998, 94: 87-97. 10.1016/S0166-6851(98)00053-X.
Montalvetti A, Pena-Diaz J, Hurtado R, Ruiz-Perez LM, Gonzalez-Pacanowska D: Characterization and regulation of Leishmania major 3-hydroxy-methyl-glutaryl -CoA reductase. Biochem J. 2000, 349: 27-34. 10.1042/0264-6021:3490027.
Pradines B, Torrentino-Madamet M, Fontaine A, Henry M, Baret E, Mosnier J, Briolant S, Fusai T, Rogier C: Atorvastatin is 10-fold more active in vitro than other statins against Plasmodium falciparum. Antimicrob Agents Chemother. 2007, 51: 2654-2655. 10.1128/AAC.01330-06.
Parquet V, Briolant B, Torrentino-Madamet M, Henry M, Almeras L, Amalvict R, Baret E, Fusaï T, Rogier , Pradines B: Atorvastatin is a promising partner for antimalarial drugs in treatment of Plasmodium falciparum malaria. Antimicrob Agents Chemother. 2009, 53: 2248-2252. 10.1128/AAC.01462-08.
Savini H, Souraud JB, Briolant S, Baret E, Amalvict R, Rogier C, Pradines B: Atorvastatin as a potential antimalarial drug: in vitro synergy in combinational therapy with dihydroartemisinin. Antimicrob Agents Chemother. 2010, 54: 966-967. 10.1128/AAC.01006-09.
Wu X, Whitfield I, Stewart BH: Atorvastatin transport in the Caco-2 cell model: contributions of P-glycoprotein and the proton-monocarboxylic acid co-transporter. Pharm Res. 2000, 17: 209-215. 10.1023/A:1007525616017.
Wang E, Casciano CN, Clement RP, Johnson WW: HMG-CoA reductase inhibitors (statins) charaterized as direct inhibitors of P-glycoprotein. Pharm Res. 2001, 18: 800-806. 10.1023/A:1011036428972.
Holtzman CW, Wiggings BS, Spinler SA: Role of P-glycoprotein in statin drug interaction. Pharmacotherapy. 2006, 26: 1601-1607. 10.1592/phco.26.11.1601.
Pradines B, Pagès JM, Barbe J: Chemosensitizers in drug transport mechanisms involved in protozoan resistance. Curr Drug Targets - Infect Disorders. 2005, 5: 411-431. 10.2174/156800505774912875.
Henry M, Alibert S, Rogier C, Barbe J, Pradines B: Inhibition of efflux of quinolines as new therapeutic strategy in malaria. Curr Top Med Chem. 2008, 8: 563-578. 10.2174/156802608783955593.
Pradines B, Parquet V, Orlandi-Pradines E: ABC Transporters in Plasmodium falciparum and their involvement in resistance to animalarial drugs. ABC transporters in microorganisms. Edited by: Ponte-Sucre A. UK: Horizon Scientific Press, 2009: 113-127.
Mu J, Ferdig MT, Feng X, Joy DA, Duan J, Furuya T, Subramanian G, Aravind L, Cooper RA, Wootton JC, Xiong M, Su XZ: Multiple transporters associated with malaria parasite responses to chloroquine and quinine. Mol Microbiol. 2003, 49: 977-989. 10.1046/j.1365-2958.2003.03627.x.
Ursing J, Zakeri S, Gil JP, Bjorkman A: Quinoline resistance associated polymorphisms in the pfcrt, pfmdr1 and pfmrp genes of Plasmodium falciparum in Iran. Acta Trop. 2006, 97: 352-356. 10.1016/j.actatropica.2006.01.002.
Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, Ursos LMB, Sidhu ABS, Naudé B, Deitsch KW, Su XZ, Wootton JC, Roepe PD, Wellems TE: Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000, 6: 861-871. 10.1016/S1097-2765(05)00077-8.
Ferdig MT, Cooper RA, Mu J, Deng B, Joy DA, Su XZ, Wellems TE: Dissecting the loci of low-level quinine resistance in malaria parasites. Mol Microbiol. 2004, 52: 985-997. 10.1111/j.1365-2958.2004.04035.x.
Henry M, Briolant S, Zettor A, Pelleau S, Baragatti M, Baret E, Mosnier J, Amalvict R, Fusai T, Rogier C, Pradines B: Plasmodium falciparum Na+/H+ exchanger 1 transporter is involved in reduced susceptibility to quinine. Antimicrob Agents Chemother. 2009, 53: 1926-1930. 10.1128/AAC.01243-08.
Lambros C, Vanderberg JP: Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979, 65: 418-420. 10.2307/3280287.
Bogreau H, Renaud F, Bouchiba H, Durand P, Assi SB, Henry MC, Garnotel E, Pradines B, Fusai T, Wade B, Adehossi E, Parola P, Kamil MA, Puijalon O, Rogier C: Genetic diversity and structure of African Plasmodium falciparum populations in urban and rural areas. Am J Trop Med Hyg. 2006, 74: 953-959.
Henry M, Diallo I, Bordes J, Ka S, Pradines B, Diatta B, M'Baye PS, Sane M, Thiam M, Gueye PM, Wade B, Touze JE, Debonne JM, Rogier C, Fusai T: Urban malaria in Dakar, Senegal: chemosusceptibility and genetic diversity of Plasmodium falciparum isolates. Am J Trop Med Hyg. 2006, 75: 146-151.
Biot C, Chavain N, Dubar F, Pradines B, Trivelli X, Brocard J, Forfar I, Dive D: Structure-activity relationship of 4-N-substituted ferroquine analoges: Time to re-evaluate the mechanism of action of ferroquine. J Organomet Chem. 2009, 694: 845-854. 10.1016/j.jorganchem.2008.09.033.
Canfield CJ, Pudney M, Gutteridge WE: Interactions of atovaquone with other antimalarial drugs against Plasmodium falciparum in vitro. Exp Parasitol. 1995, 80: 373-381. 10.1006/expr.1995.1049.
Borek-Dohalsky V, Huclova J, Barrett B, Nemec B, Ulc I, Jelinek I: Validated HPLC-MS-MS method for simultaneous determination of atorvastatin and 2-hydroatorvastatin in human plasma-pharmacokinetic study. Anal Bioanal Chem. 2006, 386: 275-285. 10.1007/s00216-006-0655-3.
Henry M, Briolant S, Fontaine A, Mosnier J, Baret E, Amalvict R, Fusai T, Fraisse L, Rogier C, Pradines B: In vitro activity of ferroquine is independent of polymprphisms in transport protein genes implicated in quinoline resistance in Plasmodium falciparum. Antimicrob Agents Chemother. 2008, 52: 2755-2759. 10.1128/AAC.00060-08.
Wurtz N, Briolant S, Gil M, Parquet V, Henry M, Baret E, Amalvict R, Almeras L, Rogier C, Pradines B: Synergy of mefloquine activity with atorvastatin, but not chloroquine and monodesethylamodiaquine, and association with the pfmdr1 gene. J Antimicrob Chemother. 2010, doi:10.1093/jac/dkq173
Bienvenu AL, Picot S: Statins alone are ineneffective in cerebral malaria but potentiate artesunate. Antimicrob Agents Chemother. 2008, 52: 4203-4204. 10.1128/AAC.00513-08.
Kobbe R, Schreiber N, May J, Jacobs T: Simvastatin treatment shows no effect on the incidence of cerebral malaria or parasitemia during experimental malaria. Antimicrob Agents Chemother. 2008, 52: 1583-1584. 10.1128/AAC.01428-07.
White NJ: Preventing antimalarial drug resistance through combinations. Drug Resist Updat. 2001, 1: 3-9. 10.1016/S1368-7646(98)80208-2.
Posvar EL, Radulovic LL, Cilla DD, Whitfield LR, Sedman AJ: Tolerance and Pharmacokinetics of single-dose atorvastatin, a potent inhibitor of HMG-CoA reductase, in healthy subjects. J Clin Pharmacol. 1996, 36: 728-731.
Clark IA, Alleva LM, Mills AC, Cowden WB: Pathogenesis of malaria and clinically similar conditions. Clin Microbiol Rev. 2004, 17: 509-539. 10.1128/CMR.17.3.509-539.2004.
Kruger PS, Freir NM, Venkatesh B, Robertson TA, Roberts MS, Jones M: A preliminary study of atorvastatin plasma concentrations in critically ill patients with sepsis. Intensive Care Med. 2009, 35: 717-721. 10.1007/s00134-008-1358-3.
Escobar C, Echarri R, Barrios V: Relative safety profiles of high dose statin regiments. Vasc Health Risk Manag. 2008, 4: 525-533.
Mills EJ, Rachlis B, Wu P, Devereaux PJ, Arora P, Perri D: Primary prevention of cardiovascular mortality and events with statin treatments. J Am Coll Cardiol. 2008, 52: 1769-1781. 10.1016/j.jacc.2008.08.039.
Silva M, Matthews ML, Jarvis C, Nolan NM, Belliveau P, Malloy M, Gandhi P: Meta-analysis of drug-induced adverse events associated with intensive-dose statin therapy. Clin Ther. 2007, 29: 253-260. 10.1016/j.clinthera.2007.02.008.
Koren MJ, Feldman T, Mendes RA: Impact of high-dose atorvastatin in coronary heart disease patients age 65 to 78 years. Clin Cardiol. 2009, 32: 256-263. 10.1002/clc.20448.
Davis TME, Pongponratan E, Supanaranond W, Pukrittayakamee S, Helliwell T, Holloway P, White NJ: Skeletal muscle involvement in falciparum malaria: Biochemical ultrastructural study. Clin Infect Dis. 1999, 29: 831-835. 10.1086/520444.
Reynaud F, Mallet L, Lyon A, Rodolfo JM: Rhabdomyolysis and acute renal failure in Plasmodium falciparum malaria. Nephrol Dial Transplant. 2005, 20: 847-10.1093/ndt/gfh686.
Mishra SK, Pati SS, Mahanta KC, Mohanty S: Rhabdomyolysis in falciparum malaria - a series of twelve cases (five children and seven adults). Trop Doc. 2010, 40: 87-88. 10.1258/td.2009.090387.
Cooper RA, Ferdig MT, Su XZ, Ursos LM, Mu J, Nomura T, Fujioka H, Fidock DA, Roepe PD, Wellems TE: Alternative mutations at position 76 of the vacuolar transmembrane protein PfCRT are associated with chloroquine resistance and unique stereospecific quinine and quinidine responses in Plasmodium falciparum. Mol Pharmacol. 2002, 61: 35-42. 10.1124/mol.61.1.35.
Sidhu AB, Valderramos SG, Fidock DA: Pfmdr1 mutations contribute to quinine resistance and enhance mefloquine and artemisinin sensitivity in Plasmodium falciparum. Mol Microbiol. 2005, 57: 913-926. 10.1111/j.1365-2958.2005.04729.x.
Klokouzas A, Tiffert T, van Schalkwyk D, Wu CP, van Veen HW, Barrand MA, Hladky SB: Plasmodium falciparum expresses a multidrug resistance-associated protein. Biochem Biophys Res Commun. 2004, 321: 197-201. 10.1016/j.bbrc.2004.06.135.
Nkrumah LJ, Riegelhaupt PM, Moura P, Johnson DJ, Patel J, Hayton K, Ferdig MT, Wellems TE, Akabas MH, Fidock DA: Probing the multifactorial basis of Plasmodium falciparum quinine resistance: Evidence for a strain-specific contribution of the sodium-proton exchanger PfNHE. Mol Biochem Parasitol. 2009, 165: 122-131. 10.1016/j.molbiopara.2009.01.011.
Shah NK, Akler AP, Sem R, Susanti AI, Muth S, Maguire JD, Duong S, Ariey F, Meshnick SR, Wongsrichanalai D: Molecular surveillance for multidrug-resistant Plasmodium falciparum, Cambodia. Emerg Infect Dis. 2008, 14: 1637-1640. 10.3201/eid1410.080080.
Carrara VI, Zwang J, Ashley EA, Price RN, Stepniewska K, Barends M, Brockman A, Anderson T, McGready R, Phaiphun L, Proux S, van Vugt M, Hutagalung R, Lwin KM, Phyo AP, Preechapornkul P, Imwong M, Pukrittayakamee S, Singhasivanon P, White NJ, Nosten F: Changes in the treatment responses to artesunate-mefloquine on the Northwestern border of Thailand during 13 years of continuous deployment. PlosOne. 2009, 4: e4451-
Rogers WO, Sem R, Tero T, Chim P, Lim P, Muth S, Socheat D, Ariey F, Wongsrichanalai C: Failure of artesunate-mefloquine combination therapy for uncomplicated Plasmodium falciparum malaria in southern Cambodia. Malar J. 2009, 8: 10-10.1186/1475-2875-8-10.
Noedl H, Se Y, Schaecher K, Smith BL, Socheat D, Fukuda MM: Evidence of artemisinin-resistant malaria in western Cambodia. N Engl J Med. 2008, 359: 2619-2620. 10.1056/NEJMc0805011.
Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwrong M, Chotivanish K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NPJ, Lindegardh N, Socheat D, White NJ: Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med. 2009, 361: 455-467. 10.1056/NEJMoa0808859.
Couto AS, Kimura EA, Peres VJ, Uhrig ML, Katzin AM: Active isoprenoid pathway in the intra-erythrocytic stages of Plasmodium falciparum: presence of dolichols of 11 and 12 isoprene units. Biochem J. 1999, 341: 629-637. 10.1042/0264-6021:3410629.
Ackowledgments
This work was supported by the Direction Centrale du Service de Santé des Armées (grant no 2007 RC 32).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
VP, MH, NW, SB and BP conceived and designed the experiments. VP, JD, EB and RA performed the in vitro experiments. MH, NW, SB and MG performed the genotyping. SB, CR and BP analysed the data. NW, SB, CR and BP wrote the paper. All authors read and approved the final manuscript.
Electronic supplementary material
12936_2010_1220_MOESM1_ESM.DOC
Additional file 1: Association of atorvastatin (AVA) in vitro responses (IC50), quinine (QN), AVA + QN, and polymorphisms in the pfnhe-1, pfcrt, pfmdr1, pfmdr2 and pfmrp genes or copy number of the pfmdr2 gene of 21 strains of Plasmodium falciparum. (DOC 40 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Parquet, V., Henry, M., Wurtz, N. et al. Atorvastatin as a potential anti-malarial drug: in vitro synergy in combinational therapy with quinine against Plasmodium falciparum. Malar J 9, 139 (2010). https://doi.org/10.1186/1475-2875-9-139
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-9-139