
Abstract
Structural model of C100H79O7NS coal organic mass was obtained within density functional theory in the localized orbital basis set using the B3LYP hybrid functional. The model was compared with the known experimental data for coal of different grades and its sorption properties were studied with respect to CH4, CO2 and H2O. It has been shown that macromolecule of coal organic mass has bulk structure with a pore inside it. Interaction between coal and CH4 molecules consists of typical physical adsorption with oligomer formation on the pore border, physical adsorption with elements of chemical adsorption was also observed between coal and H2O molecules. Interaction between coal and H2O molecules included both physical and chemical adsorbion.
Similar content being viewed by others


1 Introduction
Coal is one of the basic energy materials of organic origin, it is also the source of raw materials for chemical industry. Coal is a complex heterogeneous rock, consisting both of organic and inorganic impurities, it is characterized by developed porosity and has both chemical and physical structure. As for its chemical composition, coal is a mixture of high-molecular-weight polycyclic aromatic compounds, such as benzene C6H6, toluene C6H5CH3, xylene C6H4(CH3)2, naphthalene C10H8, anthracene C14H10, pyrene C16H10 and their derivatives with high mass fraction of carbon, as well as of water and volatile substances with small amount of mineral impurities. There is a great number of works where structures of these elementary objects are studied in detail (Zhuravlev et al. 2012). Physical structure of coal is defined by its supramolecular structure, size and pore distribution. According to the X-ray structural analysis, the pore size in coal varies from 5 to 1000 Å, the maximum being 80–100 Å.
Elemental composition of coal organic mass, structure of macromolecules and nature of supramolecular structure determine the basic physical, chemical and technological properties of coal (Gyulmaliev et al. 2003). It is impossible to determine if coal is suitable for specific technological processes without taking into account physical and chemical characteristics of coal structure. One of the main problems of coal chemistry is prediction of physical and chemical properties of coal organic mass in accordance with the structural and chemical data, that is elemental, functional and fragment. Theoretical research methods play a certain role in resolving this task (Kairbekov et al. 2015).
Nowadays there is a great number (~ 150) of molecular models of coal organic mass, their review can be found in (Castro-Marcano et al. 2012; Roberts et al. 2015; Zhang et al. 2017). Along with the growth of computation capacity, computer simulation has recently made it possible to use 3D structures of coal and predict their physical and chemical properties. Equilibrium structures and infrared spectra for four typical molecular models of coal have been determined in the works (Gagarin and Gyulmaliev 2007; Arenillas et al. 2003). Due to combination of theoretical calculations with the experimental IR-spectra it is possible to suggest probable molecular structure for wet coal. It is shown in (Xiang et al. 2014) that it is possible to successfully describe interaction of C222H185N3O17S5 coal molecule with CH4, CO2 and H2O molecules using molecular dynamics method and taking into account the contribution of van der Waals forces, electrostatic interactions and hydrogen bonds to the reduction of non-bonding interactions.
Methane plays a significant role in physical processes occurring in coal that is why the study of sorption properties of coal with regards to methane is a challenging task (Cheng et al. 2017). It is shown in (Yua et al. 2017) that methane is within coal substance, it is released on its surface in a molecular form, i.e. during the desorption process methane molecules do not dissociate when they enter the solid solution in coal. The bonding of methane molecules with coal is determined by van der Waals forces, while the incorporation energy of a methane molecule into solid solution is not a constant quantity. It depends on a specific localization of a molecule: whether it is in aliphatic compound or inside a crystallite. The methane sorbed by coal is distributed among the solid solution (absorption) and surface of cracks and pores (adsorption). As coal has a very developed internal surface, the amount of adsorbed methane can equal that of the absorbed.
The study of interaction process of CO2 and H2O with coal is also of great interest (Meng et al. 2018a, b). Chemical composition of coal is complex that makes coal both hydrophilic and hydrophobic. Compared to non-polar gases CH4 and CO2, coal-water interactions are more complicated, and mainly consist of dispersion force, hydrogen bond and chemical adsorption between water and minerals. A large number of literary sources state that as H2O content in coal increases, its adsorption properties relative to CH4 and CO2 decrease. Moisture content is one of the main parameters influencing gas adsorption in coal seams (Jinxuan et al. 2018; Meng et al. 2018a, b). CO2—coal interaction is of interest because due to it we can increase the volume of methane recovered from coal seams. The injection of CO2 into coal seams does not only reduce carbon dioxide emissions, but also improves the desorption capacity of methane displacement (Wu et al. 2019).
The purpose of this research is to establish molecular 3D-structure of coal organic mass and determine possible ways of CH4, CO2 and H2O sorption using computer-based simulation methods.
2 Computational details
Both the Restricted Hartree–Fock method (RHF) and the density functional theory (DFT) were used in this work which were successfully implemented in software code Firefly (Granovsky 2019). The density functional theory (DFT) is one of the most popular and widely-used methods in computational physics and chemistry. It is a rigorous many-electron theory based not on wave function but on electron density function ρ(r), that includes the contribution of all the electrons. Different exchange–correlation functional, both local (only the ρ dependence is used) and non-local (gradient, the \(\nabla \rho\) dependence is also used), are used for calculation of many-electron systems. The DFT methods, as they one way or another include the nonlocal Hartree–Fock exchange potential, are called hybrid methods. In this case the exchange–correlation functional is usually recorded as a linear combination:
where the \(c_{HF}\) and \(c_{DFT}\) coefficients are the parameters of the specific functional. Becke’s hybrid exchange–correlation functional combined with nonlocal Lee–Yang–Parr correlation functional (LYP), B3LYP version, was used in the present work (Becke 1993):
The exchange part of the B3LYP functional includes 20% of the Hartree–Fock exchange, 8% of the Slater-Dirac exchange and 72% of the Becke’s exchange, while the correlation part includes 19% of the VWN local functional (Vosko, Wilk and Nusair) and 81% of the LYP functional, taking into account gradient corrections.
In our calculations we used the basis set TZV (Dunning 1971), which is the most complete, and the minimal basis set developed and optimized by Husinaga MINI (Huzinaga et al. 1984).
To determine the structure of methane dimer and trimer as well as to study the sorption properties of coal organic mass, it is necessary to take into account intermolecular interaction. As is well-known the traditional approaches of DFT are not good for simulation of weak dispersion interactions, for this reason one of the perspective DFT-D (Grimme et al. 2011) methods was used. This method is based on the additive scheme of energy calculation: dispersion correction term is added to the energy obtained in the DFT-D approximation (empirical dispersion correction).
The dispersion component of the energy is calculated according to the formula (Grimme et al. 2011)
where s6 = 0.418, a1 = sr,6= 0.0, s8 = 0.25, a2 = 5.65. s6 sets up correspondence for GGA and hybrid functionals to improve the correction of the repulsive behavior at short and medium distances of the exchange functional, a1 and a2 are system’s free parameters. C nAB is an average dispersion coefficient of the nth order for the atom of AB pair, RAB is their interatomic distance, while R 0 AB is the cutoff radius for the corresponding atoms. a1 and a2 free parameters were obtained from the calculations of the methane dimer, while the recommended in (Song et al. 2017) values were used for other parameters. Based on the known values for bond energy, the dispersion correction for energy and the distance between two molecules in the dimer, the a2 free parameter was determined as 7.16. At the present value the deviation for the distance between the molecules (3.7 Å) is not more than 1% of (Johnson and Becke 2006), for bond energy (Esv = − 0.58 kcal/mol) is 8% and for the dispersion correction (Edisp = − 0.69 kcal/mol) is not more than 1%.
3 Results and discussion
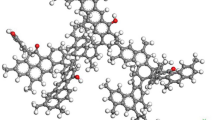
C100H79O7NS model was chosen as the basic coal organic mass (Heredy and Wender 1980). This structure contains the minimal possible number of atoms at which percentage and weight content of structural elements, characteristic of coal, is maintained, that is 83.2% of carbon, 5.5% of hydrogen, 7.7% of oxygen, 1.1% of nitrogen and 2.2% of sulfur. The aromaticity coefficient of this structure is 0.7. Besides, this structure has a good agreement with the known data on the aromatic oxygen content in coal (2.22%) and on the content of such an important structure fragment as CH3 (1.04%). In (Heredy and Wender 1980) C100H79O7NS is reported as flat and it is of illustrative character. 2D and 3D models have not been constructed on this basis before. The structures of all fragments in the C100H79O7NS:C12H9N (a), C16H10S (b), C18H12O (c) and C16H18 (d) (Fig. 1) compound have been determined to construct the 3D molecular model. Typical bond lengths and angles have been calculated and energy and frequency characteristics have been obtained. From the reported structure fragments (a) and (b) are flat structures, while (c) and (d) are bulk structures. In C18H12O molecule one benzene ring lies at an angle of 24° to the plane of the other ring. The benzene ring itself does not undergo deformations and the internal angles in it are 120°. C16H18 molecule acquires volume from the attached methyl groups. The internal angle for benzene ring remains the same only in the centre of this molecule, while in the rings, where methyl groups are attached, the angles change. In certain cases the deviation may be up to 10°. It should be noted that structural deformation also takes place in plain structures (a, b) when sulphur and nitrogen are attached, as evidenced by change in the internal angles in these molecules by 1°–2°. Two characteristic angle values are distinguished during the geometry analysis of parent molecules. They are C–N–C (a) angle equal to 110° and C–S–C angle equal to 89°. Thus, the benzene rings and their elements, to which both separate nitrogen (N) and sulphur (S) atoms and atoms of –CH2, –CH3 and –CH2–CH2–CH2– moities are directly attached, undergo the greatest deformation and change.

Gradual process of structural fragments modeling
Atoms charges have been obtained for all the reported structure fragments. Thus, the standard for the benzene ring is the Mulliken positive charge on hydrogen about + 0.1 e (e—is an electron charge) and negative charge on carbon about − 0.1 e. The sulphur atom acquires +0.18 e positive charge in C16H10S (b) molecule, while the closest carbons acquire − 0.31 e charge. As for C12H9N (a) molecule, the nitrogen atom acquires − 0.4 e negative charge, however, directly attached to it carbon atoms have a weak positive charge (0.04 e). The oxygen atom in C18H12O molecule is negative − 0.3 e, while the two attached to it carbon atoms are positive (+ 0.06 e and + 0.01 e, respectively).
To construct 3D model of coal organic mass, –CH–, –CH–CH2–, –CH2– and –O– moities were used as bridge structures between a–d molecules. The structure fragments were linked to each other at certain angles. The model of coal organic mass structure is shown in (Fig. 2) with two characteristic angles between (c) and (d) molecules and (a) and (c) molecules, which are designated as α and β, respectively. α angle is 85.7°, β angle is 114.4°. The linear sizes of the coal organic mass model presented in (Fig. 2) are 22.22 Å, 12.46 Å and 13.90 Å along x, y, z axes, respectively. The volume of van der Waals is 2011.94 Å3. Deformation of the benzene rings can be observed in the structure due to C-H bond change by 0.03 Å and C–C bond change by 0.05 Å in comparison with elemental structural fragments like naphthalene and anthracene (Zhuravlev et al. 2012). Due to connection of structure fragments by a-d bridge bond at different angles, the bulk structure of coal organic mass with “empty” space inside similar to a pore (Fig. 2b) was obtained. This pore can contain other molecules, for example the methane molecule (Fig. 2a).

Coal organic mass model
We calculated the IR spectrum of our model and carried out the Lorentz broadening of discrete spectral lines to perform a comparative analysis of the obtained structure of coal organic mas with a coal sample. By comparing the calculated IR-spectrum of coal organic mass and experimental IR-spectrum for DG coal grade (long flame) (Fig. 3), it can be stated that the obtained structure may be a part of coal structure, as evidenced by coincidence of a number of characteristic peaks. Certain characteristic regions of O–H (3300–3500 cm−1), C-H (3100–3200 cm−1) and C–O (1100–1200 cm−1) vibrations can be noted among the peaks.

a IR-spectrum of 3D model of coal organic mass, b experimental IR-spectrum for DG coal grade (long flame)
The volume (104.79 Å3) of the pore indicated in (Fig. 2b) is 1.64% from the volume of the molecular model of coal organic mass. Due to this, there is enough space for up 5 CH4 methane molecules, as the volume of one such molecule is about 12 Å3. The study of methane adsorption on coal organic mass was conducted in the following way. Methane molecules were alternately placed into a pore in such a way that the distance between the methane atoms and coal organic mass atoms was larger than that needed for chemical bond formation (~ 2 Å). The “freezing” of atoms was applied for geometry optimization for those atoms that were at some distance from the atoms’ pore of coal organic mass. The simulated tests indicated that these atoms of coal organic mass do not interact with methane molecules and do not bring significant changes to the geometry. The error of bond length is 0.4%, and the error of bond angles is 1.2%. At the same time the total energy for the “frozen” and free models differs by less than 0.01%.
Physical methane adsorption is shown in (Fig. 2a). Since the distance between any methane atom and the atom of coal organic mass molecule in the optimized structure is more than 2 Å, chemical bond is not formed. The methane concentration in the obtained model is 3.24% from the total molar weight of the molecule.
The following peculiarities have been noticed while analyzing the geometry of coal organic mass model after having placed the methane molecules in the “pore”. As the methane concentration increases, the methane molecule tends to the border of the pore, accumulation of methane takes place at the edge of the pore. At this time, dimer, trimer or oligomer, depending on the amount of methane molecules in the pore, are formed. Such conclusion is drawn from the comparison of the geometry of arrangement of methane molecules in the pore with the geometry of free dimer, trimer and oligomer (Fig. 4).

Methane tetramer (distances are given in Å)
Analyzing the geometry of the coal organic mass structure and different methane concentrations in the pore, it is possible to draw a conclusion that as the methane concentration increases, the distance from the coal organic mass structure to the nearest molecule does not increase. Thus, it proves that the pore is able to maintain the obtained structure.
To provide a more detailed analysis, it is necessary to study such important energy characteristics of the obtained model as total energy and bond energy:
where n is a number of molecules placed in a pore.
The formula (5) can be applied if we assume that the methane molecules placed in the pore do not form a polymeric structure. In case the polymeric structure is formed, the following formula should be used
It turned out that one methane molecule, placed in the center of the pore, after optimization tends to the outside and is located near the surface of the molecule. In this case the bond energy is calculated according to formula (5). At n = 1, the bond energy is − 117.26 kcal/mol. The second molecule, initially placed in the centre of the pore, also comes to the surface and together with the first methane molecule forms a dimer with a typical distance between carbon atoms of 3.7 Å. The bond energy was calculated according to formula (6), at n = 2, it is − 128.39 kcal/mol. The third methane molecule comes to the surface, too, and transforms methane dimer to trimer with carbon atoms distances of 3.7 Å and 3.8 Å. The bond energy for n = 3 is − 131.98 kcal/mol. As the methane concentration continues to increase, the tendency for polymerization is preserved. With the forth added methane atom, a tetramer is formed where the distances between carbons are 3.7 Å, 3.7 Å and 3.9 Å, the energy bond is − 147.42 kcal/mol. It should be noted that in all the cases the distance between carbons in dimer, trimer or tetramer, located close to the surface of the coal organic mass macromolecule, were less than the corresponding distances in free dimer, trimer or tetramer.
Similarly, the sorption properties of the obtained model with respect to CO2 and H2O were studied. When up to two molecules of carbon dioxide (CO2) were placed in the pore, physical adsorption takes place. With concentration increase, both physical and chemical adsorption occur, which becomes apparent when one of the CO2 molecules is destroyed and oxygen atom joins coal organic mass molecule. The remaining part of the molecule joins the other CO2 molecule, C2O3 structure is formed, which is located close to the surface of the pore. The distances between atoms in the formed molecule are 1,5 Å for single chemical bond C-O and 1,2 Å for double chemical bond. Wherein the distance between any atom of C2O3 and the closest atoms of the coal organic mass model is above 2,3 Å, which indicates the absence of chemical bond between these atoms. During the study of the sorption interaction with a molecule of water, physical adsorption with elements of chemical adsorption is observed. As the concentration of H2O molecules increases, dimers are formed.
4 Conclusion
Due to the conducted calculation within DFT theory using B3LYP hybrid functional, it is possible to develop the optimal scheme to calculate the structures of polycyclic compounds and further the structure of coal organic mass with composition similar to coal. The obtained C100H79O7NS 3D molecular model is in good agreement with the known experimental data on coal. The model is volumetric (V = 3848 Å3) and porous, so it is possible to study sorption properties on its basis. The model of physical adsorption of methane in coal is predicted, with formation of such structures as dimer, trimer or oligomer takes place. The models of sorption interaction of the obtained coal organic mass with the molecules of carbon dioxide and water are determined. All the obtained oligomers are located near the surface of the macromolecule of coal organic mass and retained by it.
References
Arenillas A, Rubiera F, Pis JJ, Cuesta MJ, Iglesias MJ, Jimenez A, Suarez-Ruiz I (2003) Thermal behaviour during the pyrolysis of low rank perhydrous coals. J Anal Appl Pyrolysis 68–69:371–385
Becke AD (1993) Density—functional thermochemistry. III. The role of exact exchange. J Chem Phys. Doi 10(1063/1):464913
Castro-Marcano F, Lobodin VV, Rodgers RP, McKenna AM, Marshall AG, Mathews JP (2012) A molecular model for Illinois No. 6 Argonne Premium coal: moving toward capturing the continuum structure. Fuel 95:35–49
Cheng Y, Jiang H, Zhang X, Cui J, Song C, Li X (2017) Effects of coal rank on physicochemical properties of coal and on methane adsorption. Int J Coal Sci Technol 4(2):129–146
Dunning TH (1971) Gaussian basis functions for use in molecular calculations. III. Contraction of (10s6p) Atomic basis sets for the first-row atoms. J Chem Phys 55:716
Gagarin SG, Gyulmaliev AM (2007) Simulation of the IR Spectra of the Organic Mass of. Coal and Coal Products. Coke and Chemistry. 50(10): 275-284
Granovsky AA (2019) Firefly version 8. http://classic.chem.msu.su/gran/firefly/index.html. Accessed 31 Jan 2019
Grimme S, Ehrlich S, Goerick L (2011) Effect of the damping function in dispersion corrected density functional theory. J Comput Chem 32:1456–1465. https://doi.org/10.1002/jcc.21759
Gyulmaliev AM, Golovin GS, Gladun TG (2003) Theoretical grounds of chemistry coal. Publishing House of Moscow State Mining University, Moscow
Heredy LA, Wender I (1980) Model structure for a bituminous coal. ACS Division of Fuel Chemistry Preprints. 25:38–45
Huzinaga S, Andzelm J, Klobukowski M, Radzio-Andzelm E, Sakai Y, Tatewaki H (1984) Gaussian basis sets for molecular calculations. Elsevier, Amsterdam
Jinxuan H, Bogomolova AK, Makarova EY, Zhaozhong Y, Yanjun L, Jianbao H, Xiaogang L (2018) Molecular simulation of H2O, CO2, and CH4 adsorption in coal micropores. Russ J Phys Chem B 12(4):714–724
Johnson ER, Becke AD (2006) A post-Hartree-Fock model of intermolecular interactions: inclusion of higher-order corrections. J Chem Phys 124(17):174104
Kairbekov ZK, Gyulmaliev AM, Yarkova TA, Smagulova NT, Kairbekov AZ (2015) Structure and properties of heavy organic residue from coal hydrogenation. Coke Chem 58(10):367–371
Meng F, Li X, Lv X, Li Z (2018a) CO hydrogenation combined with water-gas-shift reaction for synthetic natural gas production: a thermodynamic and experimental study. Int J Coal Sci Technol 5(4):439–451
Meng J, Zhong R, Li S, Yin F, Nie B (2018b) Molecular model construction and study of gas adsorption of Zhaozhuang coal. Energy Fuels 32:9727–9737. https://doi.org/10.1021/acs.energyfuels.8b01940
Roberts MJ, Everson RC, Neomagus HWJP, Niekerk DV, Mathews JP, Branken DJ (2015) Influence of maceral composition on the structure, properties and behaviour of chars derived from South African coals. Fuel 142:9–20
Song Yu, Zhu Yan-ming, Li Wu (2017) Macromolecule simulation and CH4 adsorption mechanism of coal vitrinite. Appl Surf Sci 396:291–302
Wu S, Jin Z, Deng C (2019) Molecular simulation of coal-fired plant flue gas competitive adsorption and diffusion on coal. Fuel 239:87–96
Xiang J, Zeng F, Liang H et al (2014) Molecular simulation of the CH4/CO2/H2O adsorption onto the molecular structure of coal. Sci China Earth Sci 57(8):1749–1759
Yua S, Yan-ming Z, Wu L (2017) Macromolecule simulation and CH4 adsorption mechanism of coal vitrinite. Appl Surf Sci 396:291–302
Zhang Z, Kang Q, Wei S, Yun T, Yan G, Yan K (2017) Large scale molecular model construction of Xishan bituminous coal. Energy Fuels 31:1310–1317
Zhuravlev YN, Fedorov IA, Kiyamov MY (2012) First-principles study of the crystal structure and equation of state of naphthaline and anthracene. J Struct Chem 53(3):417–423
Acknowledgements
The work was supported by the Ministry of Education and Science of the Russian Federation within the scope of contract 15.3487.2017/PP.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhuravlev, Y.N., Porokhnov, A.N. Computer simulation of coal organic mass structure and its sorption properties. Int J Coal Sci Technol 6, 438–444 (2019). https://doi.org/10.1007/s40789-019-0256-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40789-019-0256-3