
Abstract
Biologics have transformed the treatment of immune-mediated inflammatory diseases such as rheumatoid arthritis (RA) and inflammatory bowel disease (IBD). Biosimilars—biologic medicines with no clinically meaningful differences in safety or efficacy from licensed originators—can stimulate market competition and have the potential to expand patient access to biologics within the parameters of treatment recommendations. However, maximizing the benefits of biosimilars requires cooperation between multiple stakeholders. Regulators and developers should collaborate to ensure biosimilars reach patients rapidly without compromising stringent quality, safety, or efficacy standards. Pharmacoeconomic evaluations and payer policies should be updated following biosimilar market entry, minimizing the risk of imposing nonmedical barriers to biologic treatment. In RA, disparities between treatment guidelines and national reimbursement criteria could be addressed to ensure more uniform patient access to biologics and enable rheumatologists to effectively implement treat-to-target strategies. In IBD, the cost-effectiveness of biologic treatment earlier in the disease course is likely to improve when biosimilars are incorporated into pharmacoeconomic analyses. Patient understanding of biosimilars is crucial for treatment success and avoiding nocebo effects. Full understanding of biosimilars by physicians and carefully considered communication strategies can help support patients initiating or switching to biosimilars. Developers must operate efficiently to be sustainable, without undermining product quality, the reliability of the supply chain, or pharmacovigilance. Developers should also facilitate information sharing to meet the needs of other stakeholders. Such collaboration will help to ensure a sustainable future for both the biosimilar market and healthcare systems, supporting the availability of effective treatments for patients.
Similar content being viewed by others
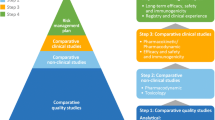

Evolution in regulatory approaches and innovation by developers are enabling price-competitive products to reach patients more quickly without compromising safety and efficacy standards. |
Pharmacoeconomic assessments need to be revisited following biosimilar market entry to remove nonmedical barriers to accessing biologic treatments, when clinically appropriate, and effective physician communication is crucial to support patient confidence in biosimilars. |
Achieving the full potential benefits of biosimilars to deliver cost savings and expanded patient access requires the efforts of, and efficient collaboration among, multiple stakeholders. |
1 Introduction
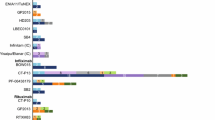
Biosimilars are biologic medicines that have no clinically meaningful differences from originator biologics (reference products [RPs]) that have already received regulatory approval. RPs have proven efficacy across many disease states and have revolutionized the treatment of immune-mediated inflammatory diseases (IMIDs), including rheumatic diseases, such as rheumatoid arthritis (RA), and inflammatory bowel disease (IBD) [1,2,3]. Multiple biosimilars have been licensed by the European Medicines Agency (EMA) and the United States (US) Food and Drug Administration (FDA) for the treatment of RA and IBD (Fig. 1), and an estimated 240 biosimilar candidates are in development across all diseases [4]. Clinical experience with biosimilars is growing, currently exceeding 700 million patient-days [5], and biosimilar uptake is increasing globally [6,7,8]. As an example, in 2017, infliximab and etanercept biosimilars held 79% and 54% of the UK market share, respectively [7].

Biologics licensed by the FDA and/or EMA for the treatment of RA and/or IBD as of 25 November 2019 (references are provided in Table S2 in the electronic supplementary material). TNF inhibitors are shown in black; biologics with other targets are shown in gray. For biologics shown in bold, biosimilars have been licensed by the FDA and/or the EMA in relevant indications. aLicensed by the FDA only. bLicensed by the FDA only for the treatment of Crohn’s disease. cThe intravenous form of golimumab (Simponi Aria®) is licensed by the FDA only, for the treatment of RA only. EMA European Medicines Agency, IBD inflammatory bowel disease, RA rheumatoid arthritis, TNF tumor necrosis factor
The reduced cost of biosimilars relative to RPs can drive market competition, contributing to budget sustainability and improving patient access to biologic treatments. Substantial cost savings attributable to biosimilars have been projected globally, with one recent estimate suggesting that direct spending on biologics could be reduced by $US54 billion in the USA from 2017 to 2026 because of biosimilars [9]. In the European Economic Area, biosimilar competition has already reduced average list prices and increased patient access to biologics [10]. Growing real-world evidence with biosimilars [11,12,13] is helping to support clinical use, increasing confidence in biosimilars. However, many patients are still treated with RPs, partly because of ongoing physician and patient concerns about biosimilars.
In this article, we aim to draw on our broad collective backgrounds to summarize our diverse, current perspectives on the future of biosimilars in IMIDs, focusing on RA and IBD. We also identify obstacles to widespread uptake of biosimilars and discuss approaches to help ensure that the benefits of biosimilars for healthcare systems and patients are maximized in the future.
2 Regulators’ and Pharmacologists’ Perspective: Rigorous and Efficient Development of Biosimilars
Regulatory approval of biosimilars is based on the totality of evidence supporting comparability of the proposed biosimilar and RP [14]. During biosimilar development, the quality attributes (structural, functional, and other analytical properties) of the RP must first be defined. The range of variation for any attribute directly impacting on the efficacy or safety of the RP (a “critical quality attribute”) must be carefully measured using multiple drug batches: these are used to define the quality target product profile of the proposed biosimilar [15,16,17]. For example, the critical quality attributes of infliximab, the RP for biosimilar CT-P13, are numerous and include those related to structure (primary and higher-order structures, glycosylation profiles), biological function (receptor-binding affinity, cytotoxicity), content (protein concentration), and impurities (host cell protein or DNA) [15].
Manufacturing processes for biosimilars must effectively be “reverse engineered” as details for RPs are not publicly available. Selection of the expression system (cell line and expression construct) is a pivotal decision as this can affect biosimilar translational and post-translational modifications and determine the number and nature of product impurities and contaminants [18]. Cell culture and product-purification processes must be gradually refined until the product possesses the target profile [15, 17]. Manufacturing processes must then be validated and carefully controlled to ensure consistent manufacture of a biosimilar candidate with this profile [15].
Demonstrating biosimilarity warrants a step-wise approach (moving from analytical analyses through nonclinical studies to clinical trials) as recommended by regulatory bodies such as the EMA and FDA [19, 20]. Analytical analyses account for most of the effort involved in biosimilar development [21, 22], with the combination of multiple orthogonal, state-of-the-art methodologies allowing a highly sensitive determination of the degree of similarity in critical quality attributes between the proposed biosimilar and the RP [14, 23]. However, current methodologies cannot provide all desirable information regarding biosimilarity at the nonclinical stage. For example, techniques to investigate how differences in post-translational modifications such as glycosylation might impact on the three-dimensional protein structure, and thus function, are lacking [24], although advances in areas such as mass spectrometry may address such issues in the future [25].
Any differences (inevitably) detected in nonclinical analyses must be demonstrated to be not clinically meaningful, with the extent and type of clinical data required for approval depending on the nature of remaining uncertainties regarding biosimilarity [20]. Therefore, unlike clinical trials for new drugs, biosimilar clinical trials are designed to establish pharmacokinetic and efficacy equivalence to the RP and to demonstrate that the proposed biosimilar and RP share similar pharmacodynamic, immunogenicity, and safety profiles [14, 17]. EMA guidelines, which are in line with FDA principles, generally recommend that biosimilar trials include a head-to-head comparison of pharmacokinetics and pharmacodynamics, followed by at least one adequately powered, randomized, parallel-group, head-to-head comparison of efficacy and safety [14, 19, 20]. Equivalence, rather than noninferiority, studies are usually required to establish that the proposed product is neither inferior nor superior to the RP [19, 20]. However, noninferiority studies may be suitable in some scenarios, such as when the RP dose is close to saturating the target [20]. Prespecified, typically symmetrical, two-sided equivalence margins or one-sided noninferiority margins must be calculated considering the variability in historical data for the RP: the margin reflects clinically acceptable differences in efficacy [26, 27]. The sample size should be adequate to detect clinically meaningful differences between biosimilar and RP and may be smaller in a noninferiority study than in an equivalence study [20]. Unlike superiority trials, per-protocol analysis is preferred for equivalence or noninferiority trials since it is more conservative than intention-to-treat analysis [28, 29].
The study population recruited into a biosimilar efficacy clinical trial should be generally aligned with one or more approved indication of the RP and should be the most sensitive for detecting any potential differences between the two drugs [19, 20]. However, while psoriasis represents a sensitive disease model to detect potential differences in clinical efficacy and immunogenicity between adalimumab biosimilars and the RP, ongoing pivotal studies more frequently include patients with RA, perhaps reflecting both patient population size and the potential commercial impact of RA [30]. In ulcerative colitis (UC), high and variable placebo response rates [31, 32] may also present a challenge for selection of this indication for biosimilar trials. The endpoints used in biosimilar clinical trials should be appropriate and sensitive enough to detect potential differences between the RP and the proposed biosimilar [19, 20]. Considering CT-P13 evaluation in IMIDs, multiple endpoints were evaluated during the PLANETRA equivalence study, including Disease Activity Score in 28 joints (DAS28), based on C-reactive protein (CRP), and American College of Rheumatology and European League Against Rheumatism (EULAR) response rates; the endpoints were highly similar between CT-P13 and reference infliximab at week 30 [33]. The postmarketing phase III PLANETCD study comparing CT-P13 and reference infliximab in patients with Crohn’s disease (CD) used a Crohn’s Disease Activity Index-based primary endpoint [34], whereas two cohort studies comparing these agents in patients with either CD or UC employed composite primary endpoints (comprising death, CD-related surgery, all-cause hospitalization, and reimbursement for other biologics) [35, 36]. The sensitive endpoints of endoscopic remission/mucosal healing were also employed in the PLANETCD study as a tertiary efficacy endpoint [34] and have been assessed in prospective observational studies of CT-P13 treatment in patients with UC [37,38,39,40,41].
Intrinsic to the clinical safety and efficacy assessment of biosimilars is evaluation of immunogenicity, as detailed in FDA and EMA guidelines [20, 42]. Anti-drug antibodies (ADAs) may affect biologic efficacy by modulating pharmacokinetics for example, or adversely affect safety, with consequences including infusion-related reactions and serum sickness [43, 44]. Multiple factors contribute to the risk of immunogenicity, including post-translational modifications such as glycosylation, as discussed in a recent review [43]. Although the authors noted that glycosylation is a common area of divergence between biosimilars and RPs, this has not led to discrepancies in immunogenicity for any EMA-approved biosimilar [43]. Immunogenicity assay development and validation must follow a rigorous process, in line with the detailed regulatory guidance [45, 46]. With multiple approaches possible, different technologies should be compared to ensure assay suitability, as conducted for CT-P13 [47]. However, debate continues regarding optimal assay design, with variations in assays used leading to extensive variability in reported immunogenicity and posing a challenge for interstudy comparability [48, 49]: for infliximab treatment, a recently published systematic review highlighted that reports of ADA-positive patients ranged from 0 to 65.3% [50].
Alongside the difficulties for immunogenicity assays, selection of appropriate pharmacodynamic markers for some biologics may be challenging because of ambiguities in their mechanism of action. For example, the complexity of tumor necrosis factor (TNF) signaling means that the mechanisms of action of TNF inhibitors are still poorly understood, particularly in IBD [51], compared with the relatively well-characterized depletion of cluster of differentiation 20-positive (CD20+) B cells underlying rituximab efficacy in RA [52]. This warrants the use of multiple pharmacodynamic markers in clinical studies evaluating TNF inhibitors, with targets, assay types, and methodologies considered on a case-by-case basis. In RA, an approach combining the evaluation of biomarkers representing immune and inflammatory processes (rheumatoid factor and anticyclic citrullinated peptide, and CRP and erythrocyte sedimentation rate, respectively [53]) may be required.
While regulatory agency guidelines on biosimilar development are broadly aligned scientifically and recommend the step-wise performance of analytical, nonclinical, and clinical studies, agencies vary in requirements and, sometimes, in their interpretations of the same data (Table 1 [54, 55]). For example, the FDA requires at least one clinical pharmacokinetic study including the version of the RP licensed in the USA, whereas Swissmedic, the Swiss regulatory agency, prefers that comparability studies use the RP from Switzerland, and Health Canada provides additional guidance for developers selecting a non-Canadian RP [20, 56, 57]. Regulatory agencies may also require studies supporting extrapolation of clinical data to patients of different ethnicities. For example, for the Japanese regulatory authority to consider efficacy and safety data from the global multicentre PLANETRA study [33], a bridging study comparing the pharmacokinetics, efficacy, and safety of CT-P13 and reference infliximab was performed in Japanese patients with RA, followed by an extension study evaluating the safety of CT-P13 during long-term treatment or after switching from reference infliximab [58, 59]. It is worth noting that geographical variations in regulatory requirements may contribute to the reluctance of some physicians to fully adopt biosimilars in clinical practice, alongside concerns surrounding indication extrapolation, as exemplified in IBD [3] and discussed later in Sects. 4 and 7.
Regulatory agencies recommend that biosimilar manufacturers align their development strategies with each agency’s specifications and hold discussions with the agencies before and during the development of each biosimilar [60,61,62] to minimize inefficiencies and delays. A robust biosimilar development plan should include strategies to ensure that, after biosimilar approval, the clinical safety of the product is appropriately monitored and any potential shortages in drug supply are prevented [63, 64]. Proper management of the biosimilar supply chain is vital to avoid product shortages, which could compromise patient safety and clinical outcomes, and might be caused by manufacturing issues, delays in supply, or other disruptions [64]. The EMA requires that descriptions of a risk management plan and pharmacovigilance system be included in any application for biosimilar approval, capturing risks and any specific monitoring associated with the RP [19, 65]. In the USA, as for any medication, the FDA may request that a risk evaluation and mitigation strategy is established for approved biosimilars [66].
Biosimilar developers should be mindful of possible future changes in approval pathways, as regulatory agencies aim to streamline and accelerate development and review processes for medical products, for example via the US 21st Century Cures Act [67, 68].
3 Rheumatologists’ Perspective: Improved Access by Addressing Disparities in Reimbursement Criteria
RA treatment aims to achieve tight disease control (long-term low disease activity or remission), thereby preventing irreversible joint damage, maximizing long-term health-related quality of life, and reducing long-term societal costs [69,70,71,72]. To accomplish this, early diagnosis and treatment are essential; however, striking the right balance between clinical benefits, risks, and economic considerations can be challenging.
Biologics have transformed the treatment of the rheumatic diseases, including RA, psoriatic arthritis, and ankylosing spondylitis (AS) [1, 2]. In recent years, RA treatment guidelines have proliferated, given the increasing cost of management in the biologic era. Current guidelines recommend access to biologics based on disease duration, disease severity, and number of insufficient responses to conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) [69,70,71,72,73,74,75,76], although differences in expert opinion may lead to divergent interpretations of the same evidence between guidelines. In addition, high costs and poor affordability often restrict patient access to biologics [77,78,79,80,81], alongside factors such as prescription controls and limitations in access to healthcare services [81]. Furthermore, national reimbursement criteria can be more restrictive than treatment guidelines, leading to disparities in patient access to biologics among countries [77, 82]. For example, a recent study in the European region (defined as 37 European countries, plus Russia and Turkey) found that only 59% of patients eligible for biologics according to EULAR guidelines (DAS28 > 3.2 and ≥ 2 csDMARD treatment failures) remained eligible following application of national reimbursement criteria [79]. The analysis also found that 16 of 39 countries required DAS28 > 5.1 for reimbursement [79], restricting biologics to patients with high disease activity and limiting the ability of rheumatologists to effectively implement a treat-to-target approach. Indeed, persistently moderate elevation of DAS28 has been associated with functional deterioration in some patients [83], whereas evidence confirming the efficacy of TNF inhibitors in patients with moderate disease activity (DAS28 > 3.2 to ≤ 5.1) is emerging from studies solely evaluating this population [84]. In general, low socioeconomic status of a country is linked to stricter reimbursement criteria, although UK criteria are notably more restrictive than this would suggest [77, 80,81,82]. Biosimilars present the opportunity for more cost-effective biologic treatment, which should help to address the disparities in patient access imposed by stringent national reimbursement criteria that may diverge from clinical guidelines.
4 Gastroenterologists’ Perspective: Earlier Introduction of Biologics for Better Clinical Outcomes
IBD treatment aims to change the natural history of the disease, thereby avoiding tissue damage and disability [85]. This requires prompt, early treatment and implementation of a treat-to-target approach (top-down or accelerated step-up) to achieve objective endpoints such as endoscopic remission [86,87,88,89,90,91]. Consequently, treatment paradigms are changing to introduce biologics and/or immunomodulators at an earlier stage of disease [87, 90, 92,93,94,95]. Numerous guidelines address the role of biologics in IBD treatment [96,97,98,99,100,101,102], generally recommending biologics as a treatment option for steroid-refractory/dependent disease or in patients who are intolerant of, or have contraindications to, conventional therapy. Early treatment with biologics is recommended in certain patient populations, including those with a poor prognosis or extensive or severely active disease [96, 97, 103].
A “window of opportunity” exists in patients with CD, during which intervention with disease-modifying anti-IBD drugs should occur [104] and TNF inhibitor treatment can be much more effective than later use [93]. Available data on early IBD treatment with TNF inhibitors are summarized in Table S1 in the electronic supplementary material. For infliximab, studies in pediatric patients have reported that early treatment is associated with improved clinical outcomes, including a reduced rate of relapse and increased mucosal healing [105,106,107]. The CHARM trial in adalimumab-treated patients with moderate-to-severe CD found the highest remission rates and lowest side effects in the shortest disease duration subgroup (< 2 years) [108]. Early treatment escalation to adalimumab in the CALM trial resulted in a significantly higher proportion of patients achieving endoscopic, steroid-free, and clinical remission versus clinical management, with a similar incidence of treatment-emergent adverse events [109]. Early versus late treatment is also associated with improved outcomes in retrospective analyses of TNF inhibitor-treated patients with CD [110,111,112,113]. Benefits of early vedolizumab treatment have also been described [114,115,116].
Biosimilars were first licensed for IBD treatment in 2013, with EMA approval of CT-P13 across all infliximab indications [117]. Several infliximab and adalimumab biosimilars have subsequently been approved by the EMA and the FDA, with more in the pipeline [118, 119]. To date, approval of biosimilars for the treatment of IBD has been based on “extrapolation,” where approval is permitted in licensed indications of the RP beyond those evaluated in biosimilar clinical trials [3]. For extrapolation to be permitted, biosimilarity must have been demonstrated in in vitro, nonclinical, and clinical studies, and the rationale for extrapolation must be scientifically justifiable [3]. Today, approval of CT-P13 in IBD is also supported by real-world evidence and clinical trial data [34, 120,121,122,123], including from the randomized, double-blind, noninferiority phase IV NOR-SWITCH study [120] and a Hungarian prospective observational cohort study [124]. To our knowledge, no published clinical trials evaluating the approved adalimumab biosimilars ABP 501, BI 695501, or SB5 in IBD, or real-world evidence for other infliximab biosimilars exist. All three adalimumab biosimilars have demonstrated pharmacokinetic similarity to the RP in equivalence studies in healthy subjects [125,126,127] and similar efficacy and safety in clinical trials in patients with RA [128,129,130].
5 Pharmacoeconomists’ Perspective: Cost of and Access to Biologic Therapy Reframed by Biosimilars
Biologics represent a significant cost for both RA and IBD treatment, with high costs limiting patient access. However, patent expiration of biologics has enabled development of lower-cost biosimilars, leading to increased market competition and price reductions [7]. In RA, initial pharmacoeconomic analyses suggested poor cost-effectiveness of TNF inhibitors versus csDMARDs in patients naïve or with an insufficient response to csDMARDs [131]. However, these analyses were performed before biosimilar market entry, missing the impact of biosimilars on price competition and consequent reduced treatment costs and expanded patient access [132]. Biosimilars, market competition, and price reductions improve the cost-effectiveness of biologic therapy in RA. In IBD, existing data support the potential for biosimilars to deliver cost savings and improve patient access to biologics [133, 134], and cost-effectiveness analyses are expected to be continuously updated. The cost-effectiveness of early biologic treatment in IBD is also being explored [135], whereas a model extrapolating data from the CALM trial in CD showed that “tight control,” using biomarkers to direct adalimumab treatment, was cost-effective versus standard clinical management over 2- and 5-year timeframes, with cost-effectiveness improving over time [136].
Biosimilar uptake may also bring indirect economic benefits, often not considered in cost-effectiveness analyses [137]. In RA, effective early treatment with DMARDs [138] or biologics [139] can improve workforce productivity, with economic benefits for individuals and society. Future cost-effectiveness analyses should endeavor to be as accurate as possible, incorporating the most relevant information, and be updated regularly; the importance of revisiting economic analyses following oncology biosimilar availability has been noted [140]. Inaccurate pricing information could lead to unfavorable cost-effectiveness estimates, restricting patient access to treatment.
Budget impact analyses have predicted that biosimilar uptake could generate significant cost savings, reduce the cost of illness, and increase patient access [141, 142]. One analysis suggested that the introduction of CT-P13 in six European countries could deliver net savings of €15.3 million over 3 years, increasing to €20.8 million if 80% of infliximab-treated patients also switched [143]. Such cost savings must be balanced against potential budget increases from expanding patient access to biologics and providing additional services for patients [78, 144]: in the aforementioned analysis, the cost savings from CT-P13 uptake could be used to treat an additional 1200–1800 patients over 3 years [143]. Cost savings may also enable earlier access to biologics in some countries [145, 146]. If cost savings were directed toward additional services, therapeutic drug monitoring (TDM) could help optimize TNF inhibitor therapy and deliver personalized care for patients with IBD [147, 148]. TDM involves measurement of drug and ADA levels: ADA formation is frequently associated with reduced primary efficacy and loss of response to IBD treatment [50] and may be predicted by factors including drug clearance and body weight [149]. Proactive TDM may be beneficial for longer-term outcomes [150]. However, the TAXIT and TAILORIX trials did not identify any benefit on clinical remission rates, although factors including the high incidence of dose intensification in the TAILORIX clinical care group may explain this [151, 152]. Currently, proactive TDM is performed in limited numbers of patients with IBD receiving TNF inhibitors [147] and is not widely recommended unless likely to impact on clinical management, although cost-effectiveness analyses demonstrate its benefits [153]. In addition, the increased affordability of biosimilars relative to biologics could facilitate clinical research incorporating these agents.
Predicted cost savings from biosimilar uptake are borne out in real-world experience. In the 2017–2018 financial year, the UK National Health Service saved £324 million through switching to biosimilars or generics for ten medicines, with almost £100 million saved through uptake of infliximab biosimilars [154]. Similarly, an analysis of rheumatology specialties in the UK (2014–2017) found that £38.8 million was saved over 2 years following the introduction of infliximab and etanercept biosimilars, because of biosimilar uptake and RP price reductions [155]. In Scandinavian countries, biosimilar availability has heralded significant cost savings and expanded patient access to biologics, despite already high use [10, 132]. In Norway, most patients with newly diagnosed IBD have received biosimilar infliximab since 2014, and, more recently, patients receiving infliximab maintenance therapy have switched to biosimilar infliximab [156]: following the introduction of infliximab biosimilars, patient access increased and expenditure reduced [132]. A mandatory switch to biosimilar infliximab for IBD induction and maintenance therapy has also been partially implemented in Norway, Poland, and the UK [157], and France aims to reach 80% biosimilar penetration by 2022, partly through switching RP-treated patients [158].
Evidence suggests no safety or efficacy concerns with a single switch from RP to biosimilar in IBD or RA [12, 34, 120, 159, 160]. For example, in biologic-naïve patients with CD, safety and efficacy were similar over 54 weeks for patients receiving reference infliximab throughout, CT-P13 throughout, or switching to the other agent at week 30 [34]. Furthermore, there was no clinically meaningful difference in the proportion of ADA-positive patients between groups; the proportion of neutralizing antibody-positive patients was also similar [34]. However, data regarding the economic impact and healthcare resource use of nonmedical switching are limited, as highlighted in a recent systematic literature review that identified few studies using real-world estimations and methodological shortcomings in others [161]. Any increase in direct financial or healthcare professional (HCP) time costs related to switching patients could present an obstacle to biosimilar uptake. Data from the DANBIO registry of patients with inflammatory rheumatic disease demonstrated that a mandatory switch from reference infliximab to CT-P13 resulted in only minor changes to outpatient healthcare resource use [11], whereas a recent study found that the modest cost savings offered by biosimilar etanercept did not justify the additional workload involved in actively switching patients from the RP in some Swedish counties [162]. Data regarding single switches are accumulating, but, to our knowledge, no published studies have investigated cross-switching (between two biosimilars) or multiple/repeated switches in RA or IBD [159, 163, 164] with regard to efficacy, safety, or cost considerations. However, evidence regarding multiple switching is emerging in psoriasis, with no impact on efficacy, safety, or immunogenicity detected after up to four switches between reference adalimumab and GP2017 [165] or up to three switches between reference etanercept and GP2015 [166]. Nevertheless, future studies should cautiously assess multiple and cross-switches in patients with other IMIDs; the pharmacoeconomic implications remain to be determined.
6 Patients’ Perspective: Improved Information Sharing May Encourage Greater Acceptance of Biosimilars
Successful biosimilar uptake requires that patients understand the rationale for, and have any concerns allayed about, initiating or switching to biosimilars. Indeed, the European Crohn’s and Colitis Organisation (ECCO) advocates that patients must be fully informed to allow evidence-based patient choice, and that such communication must consider patient health literacy [159]. ECCO notes that HCPs must transparently communicate “the tangible benefits of the biosimilar product” to patients, and that nurses can play a crucial role [159, 167]. However, reported patient awareness and understanding of biosimilars varies. A 2014–2015 European survey of 1059 patients with IBD found that approximately 36% had heard of biosimilars [168], and 49% of Belgian patients with RA (surveyed in 2016) reported familiarity with biosimilars [169]. In a 2017 UK survey of patients with RA and AS, 66% and 80% of respondents receiving RPs or biosimilars, respectively, understood what biosimilars were [170]. Among European patients with IBD who had heard of biosimilars, safety and efficacy were the most common concerns (47% and 39%, respectively), whereas around one-quarter of respondents had no specific concerns [168]. Furthermore, patients with autoimmune conditions expressed concerns that nonmedical switching could influence treatment outcomes [171].
Patients may experience nocebo effects (worsening or incitement of symptoms that are induced by a negative attitude toward an intervention) [172] that are only perceptible to the patient [173] and may impact on quality of life, treatment adherence, and the cost-saving potential of biosimilars [172, 174]. In a study of 100 patients with RA, AS, or psoriatic arthritis, 89% initially accepted a nonmedical switch from reference infliximab to CT-P13 [175]. However, 28% of patients requested a re-switch to reference infliximab, 44% of whom had no worsening of disease activity, suggesting that negative perceptions about biosimilars affected retention rates [175]. Another study of 125 patients with IBD or rheumatic disease found that 12.8% experienced a nocebo response following a nonmedical switch from RP to biosimilar infliximab [176]; in the DANBIO registry, subjective reasons mainly accounted for back-switching from biosimilar to reference etanercept [13]. Nocebo responses to biosimilars may be triggered by multiple interacting patient-related factors and psychological mechanisms influenced by both information provided to patients and the therapeutic environment [174]. Experiences shared by patients, as well as media coverage and results of internet searches, may also influence perceptions of biosimilars, contributing to nocebo effects [174]. Considering nocebo effects in patient–HCP communication may help to minimize the nocebo effect experienced and improve outcomes [174]. HCPs can minimize biosimilar-related nocebo effects through being well-informed and confident, facilitating shared, informed decision making with patients [177]. Positive framing, contextualized informed consent, and unified communications can also reduce nocebo effects [172, 174]. Despite the increasing use of internet health information by patients, one survey found that perceived physician quality had a greater impact on treatment compliance than the perceived quality of internet health information [178], emphasizing the importance of HCP communication. In addition, educational materials developed by medical societies or government organizations, in conjunction with patient groups, could play a key role in supporting patient education about biosimilars, as recognized in the oncology setting [179].
7 Developers’ Perspective: Innovative Approaches to Bring Additional Value
Developers must minimize concerns about clinical outcomes and long-term performance of biosimilars through the generation, publication, and distribution of data appropriate for stakeholders, including physicians, patients, and payers, beyond that required for regulatory approval [180,181,182]. Educational activities and publication of data in respected medical journals can help to build trust and familiarity with biosimilars among physicians [183]. For example, an evidence-based approach helped to alleviate physicians’ concerns about indication extrapolation and switching from reference infliximab to CT-P13 in the treatment of IBD [184, 185], perhaps contributing to a change in ECCO’s position on biosimilars [159]. Additional analyses of clinical data, reflecting medical society guidelines, can be conducted to support biosimilar use. For example, efficacy analyses in different patient subgroups and analysis populations, and implementation of alternative statistical techniques for handling missing data, were reported from the extension phases of the CT-P13 PLANETRA and PLANETAS studies [160, 186]. To aid the understanding of some stakeholders, including patients and payers, publications can be translated into local languages or data can be effectively communicated using lay summaries.
As more biosimilars arrive on the market, competition will push prices down [7]. Developers and payers should align pricing and market access policies to support a sustainable biosimilar market, where developers’ investments are balanced by a fair price and healthy competition is encouraged [144]. Companies must be efficient, not only at the production level but across the entire process from research and development to distribution, to maintain sustainability in the long term. Beneficially, the ability to extrapolate the clinical use of biosimilars to other indications minimizes drug development and regulatory approval costs [117]. Gains from biosimilar development could also be extended through the use of innovative approaches, such as the development of more convenient or longer-acting drug formulations, as conducted by RP manufacturers [7]. For example, infliximab biosimilars are currently administered intravenously, but a subcutaneous formulation of CT-P13 that could offer increased convenience for patients is in development [187,188,189]. Adding value to a product and dedication to an evidence-based approach will be key factors in determining the future success and sustainability of a developer in the competitive biosimilars market.
8 Conclusions
The cost savings generated by biosimilar uptake could enable improved patient access to biologic treatments for those in need. However, to maximize gains from biosimilar uptake, each stakeholder must fulfill their responsibilities as efficiently as possible. While the advent of biosimilars required regulators to adopt new frameworks, past and ongoing evolution in their scientific approach to biosimilar regulation is helping to minimize development time and investment required for biosimilar approval. This enables biosimilars to reach patients more quickly, without compromising safety and efficacy standards. Payers should revisit pharmacoeconomic assessments to ensure they reflect the impact of biosimilar market entry to relieve nonmedical barriers to biologic treatment for patients. Physicians should improve their understanding of biosimilars, increasing their confidence in prescribing biosimilars in line with treatment and healthcare funder guidelines to maximize cost savings. Patients and physicians must recognize the potential for nocebo effects. Physician communication strategies should be improved to limit the impact of nocebo effects on clinical outcomes and treatment discontinuation, to improve patient experiences, and to realize the cost-saving potential of biosimilars. Developers must be efficient to minimize costs and be price competitive without undermining product quality, the sustainability of supply, or pharmacovigilance systems. Additional innovation might be required for a product to be competitive in the context of multiple approved biosimilars. Developers should diligently provide data to meet the requirements of each stakeholder. In summary, all stakeholders must collaborate efficiently to realize the ultimate goal of biosimilar development: delivering the clinical benefits of biologic therapy to patients without sacrificing the sustainability of the healthcare system.
Change history
02 June 2020
The affiliation of the corresponding author Silvio Danese, which currently reads.
References
Rein P, Mueller RB. Treatment with biologicals in rheumatoid arthritis: an overview. Rheumatol Ther. 2017;4:247–61.
Sarzi-Puttini P, Ceribelli A, Marotto D, et al. Systemic rheumatic diseases: from biological agents to small molecules. Autoimmun Rev. 2019;18:583–92.
Ben-Horin S, Vande Casteele N, Schreiber S, et al. Biosimilars in inflammatory bowel disease: facts and fears of extrapolation. Clin Gastroenterol Hepatol. 2016;14:1685–96.
Kent D, Rickwood S, and Di Biase S. Disruption and maturity: the next phase of biologics. 2017; https://www.iqvia.com/-/media/iqvia/pdfs/nemea/uk/disruption_and_maturity_the_next_phase_of_biologics.pdf?_=1518214264988. Accessed 17 Dec 2019.
van den Hoven A. Biosimilar medicines clinical use: an experience-based EU perspective. 2017; https://www.medicinesforeurope.com/docs/20170713%20-%20Biosimilar%20Medicines%20Group,%20EU%20experience-AVH-US%20FDA%20Adcom.pdf. Accessed 17 Dec 2019.
IMS Institute for Healthcare Informatics. Delivering on the potential of biosimilar medicines. The role of functioning competitive markets. 2016; https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/delivering-on-the-potential-of-biosimilar-medicines.pdf. Accessed 17 Dec 2019.
IQVIA. The impact of biosimilar competition in Europe. 2018; https://ec.europa.eu/docsroom/documents/31642. Accessed 17 Dec 2019.
Jacoby R, Smith E, Wilkins D, et al. Winning with biosimilars. Opportunities in global markets. 2016; https://www2.deloitte.com/content/dam/Deloitte/us/Documents/life-sciences-health-care/us-lshc-biosimilars-whitepaper.pdf. Accessed 17 Dec 2019.
Mulcahy AW, Hlavka JP, and Case SR. Biosimilar cost savings in the United States: initial experience and future potential. 2017; https://www.rand.org/pubs/perspectives/PE264.html. Accessed 17 Dec 2019.
QuintilesIMS. The impact of biosimilar competition in Europe. 2017; http://ec.europa.eu/growth/content/impact-biosimilar-competition-price-volume-and-market-share-update-2017-0_en. Accessed 17 Dec 2019.
Glintborg B, Sorensen J, Hetland ML. Does a mandatory non-medical switch from originator to biosimilar infliximab lead to increased use of outpatient healthcare resources? A register-based study in patients with inflammatory arthritis. RMD Open. 2018;4:e000710.
Glintborg B, Sørensen IJ, Loft AG, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76:1426.
Glintborg B, Loft AG, Omerovic E, et al. To switch or not to switch: results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Ann Rheum Dis 2019;78:192.
Al-Sabbagh A, Olech E, McClellan JE, et al. Development of biosimilars. Semin Arthritis Rheum. 2016;45:S11–8.
Vulto AG and Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology (Oxford) 2017;56:iv14–29.
Tsuruta LR, Lopes dos Santos M, and Moro AM. Biosimilars advancements: moving on to the future. Biotechnol Prog 2015;31:1139–49.
Isaacs J, Goncalves J, Strohal R, et al. The biosimilar approval process: how different is it? Consider Med. 2017;1:3–6.
US Food and Drug Administration. Development of therapeutic protein biosimilars: comparative analytical assessment and other quality-related considerations. Guidance for industry. 2019; https://www.fda.gov/downloads/drugs/guidances/ucm291134.pdf. Accessed 17 Dec 2019.
European Medicines Agency. Guideline on similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. 2014; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/01/WC500180219.pdf. Accessed 17 Dec 2019.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. 2015; https://www.fda.gov/downloads/drugs/guidances/ucm291128.pdf. Accessed 17 Dec 2019.
Declerck P and Farouk Rezk M. The road from development to approval: Evaluating the body of evidence to confirm biosimilarity. Rheumatology (Oxford) 2017;56:iv4–iv13.
Bui LA, Hurst S, Finch GL, et al. Key considerations in the preclinical development of biosimilars. Drug Discov Today. 2015;20(Suppl 1):3–15.
Weise M, Bielsky M-C, De Smet K, et al. Biosimilars: what clinicians should know. Blood. 2012;120:5111–7.
Berkowitz SA, Engen JR, Mazzeo JR, et al. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11:527–40.
Háda V, Bagdi A, Bihari Z, et al. Recent advancements, challenges, and practical considerations in the mass spectrometry-based analytics of protein biotherapeutics: a viewpoint from the biosimilar industry. J Pharm Biomed Anal. 2018;161:214–38.
United States Food & Drug Administration. Guidance for industry: E9 statistical principles for clinical trials. 1998; https://www.fda.gov/media/71336/download. Accessed 17 Dec 2019.
European Medicines Agency. Note for guidance on statistical principles for clinical trials. 1998; https://www.ema.europa.eu/en/documents/scientific-guideline/ich-e-9-statistical-principles-clinical-trials-step-5_en.pdf. Accessed 17 Dec 2019.
US Food and Drug Administration. Non-inferiority clinical trials to establish effectiveness. 2016; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/non-inferiority-clinical-trials. Accessed 17 Dec 2019.
Alten R, Cronstein BN. Clinical trial development for biosimilars. Semin Arthritis Rheum. 2015;44:S2–8.
Lai Z, La Noce A. Key design considerations on comparative clinical efficacy studies for biosimilars: adalimumab as an example. RMD Open. 2016;2:e000154.
Jairath V, Zou GY, Parker CE, et al. Placebo response and remission rates in randomised trials of induction and maintenance therapy for ulcerative colitis. Cochrane Database Syst Rev 2017;9:Cd011572.
Ma C, Guizzetti L, Panaccione R, et al. Systematic review with meta-analysis: endoscopic and histologic placebo rates in induction and maintenance trials of ulcerative colitis. Aliment Pharmacol Ther. 2018;47:1578–96.
Yoo DH, Hrycaj P, Miranda P, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72:1613–20.
Ye BD, Pesegova M, Alexeeva O, et al. Efficacy and safety of biosimilar CT-P13 compared with originator infliximab in patients with active Crohn’s disease: an international, randomised, double-blind, phase 3 non-inferiority study. Lancet. 2019;393:1699–707.
Meyer A, Rudant J, Drouin J, et al. Effectiveness and safety of reference infliximab and biosimilar in Crohn disease: a French equivalence study. Ann Intern Med. 2019;170:99–107.
Meyer A, Rudant J, Drouin J, et al. The effectiveness and safety of infliximab compared with biosimilar CT-P13, in 3112 patients with ulcerative colitis. Aliment Pharmacol Ther. 2019;50:269–77.
Kolar M, Duricova D, Bortlik M, et al. Infliximab biosimilar (Remsima™) in therapy of inflammatory bowel diseases patients: experience from one tertiary inflammatory bowel diseases centre. Dig Dis. 2017;35:91–100.
Farkas K, Rutka M, Balint A, et al. Efficacy of the new infliximab biosimilar CT-P13 induction therapy in Crohn’s disease and ulcerative colitis—experiences from a single center. Expert Opin Biol Ther. 2015;15:1257–62.
Farkas K, Rutka M, Golovics PA, et al. Efficacy of infliximab biosimilar CT-P13 induction therapy on mucosal healing in ulcerative colitis. J Crohns Colitis. 2016;10:1273–8.
Balint A, Rutka M, Kolar M, et al. Infliximab biosimilar CT-P13 therapy is effective in maintaining endoscopic remission in ulcerative colitis—results from multicenter observational cohort. Expert Opin Biol Ther. 2018;18:1181–7.
Tursi A, Allegretta L, Chiri S, et al. Effectiveness and safety of infliximab biosimilar CT-P13 in treating ulcerative colitis: a real-life experience in IBD primary centers. Minerva Gastroenterol Dietol. 2017;63:313–8.
European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies—non-clinical and clinical issues. 2014; http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/06/WC500128686.pdf. Accessed 17 Dec 2019.
Doevendans E, Schellekens H. Immunogenicity of innovative and biosimilar monoclonal antibodies. Antibodies (Basel, Switzerland). 2019;8:21.
Jani M, Dixon WG, Chinoy H. Drug safety and immunogenicity of tumour necrosis factor inhibitors: the story so far. Rheumatology (Oxford). 2018;57:1896–907.
US Food and Drug Administration. Immunogenicity testing of therapeutic protein products—developing and validating assays for anti-drug antibody detection. 2019; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/immunogenicity-testing-therapeutic-protein-products-developing-and-validating-assays-anti-drug. Accessed 17 Dec 2019.
European Medicines Agency. Guideline on immunogenicity assessment of therapeutic proteins. 2017; https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf. Accessed 17 Dec 2019.
Kim JS, Kim SH, Kwon B, et al. Comparison of immunogenicity test methods used in clinical studies of infliximab and its biosimilar (CT-P13). Expert Rev Clin Immunol. 2015;11:33–41.
Schreitmüller T, Barton B, Zharkov A, et al. Comparative immunogenicity assessment of biosimilars. Future Oncol. 2019;15:319–29.
Gorovits B, Baltrukonis DJ, Bhattacharya I, et al. Immunoassay methods used in clinical studies for the detection of anti-drug antibodies to adalimumab and infliximab. Clin Exp Immunol. 2018;192:348–65.
Vermeire S, Gils A, Accossato P, et al. Immunogenicity of biologics in inflammatory bowel disease. Therap Adv Gastroenterol 2018;11:1756283x17750355.
Billmeier U, Dieterich W, Neurath MF, et al. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol. 2016;22:9300–13.
Cohen MD, Keystone E. Rituximab for rheumatoid arthritis. Rheumatol Ther. 2015;2:99–111.
Nakken B, Papp G, Bosnes V, et al. Biomarkers for rheumatoid arthritis: from molecular processes to diagnostic applications-current concepts and future perspectives. Immunol Lett. 2017;189:13–8.
Cazap E, Jacobs I, McBride A, et al. Global acceptance of biosimilars: importance of regulatory consistency, education, and trust. Oncologist. 2018;23:1188–98.
Kirchhoff CF, Wang XM, Conlon HD, et al. Biosimilars: key regulatory considerations and similarity assessment tools. Biotechnol Bioeng. 2017;114:2696–705.
Swissmedic. Guidance document authorisation biosimilar HMV4. 2019; https://www.swissmedic.ch/swissmedic/en/home/legal/legal-basis/administrative-ordinances.html. Accessed 17 Dec 2019.
Health Canada. Guidance document: information and submission requirements for biosimilar biologic drugs. 2016; https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs-1.html. Accessed 17 Dec 2019.
Takeuchi T, Yamanaka H, Tanaka Y, et al. Evaluation of the pharmacokinetic equivalence and 54-week efficacy and safety of CT-P13 and innovator infliximab in Japanese patients with rheumatoid arthritis. Mod Rheumatol. 2015;25:817–24.
Tanaka Y, Yamanaka H, Takeuchi T, et al. Safety and efficacy of CT-P13 in Japanese patients with rheumatoid arthritis in an extension phase or after switching from infliximab. Mod Rheumatol. 2017;27:237–45.
European Medicines Agency. Tailored scientific advice to support step-by-step development of new biosimilars. News 16/12/2016; https://www.ema.europa.eu/en/news/tailored-scientific-advice-support-step-step-development-new-biosimilars. Accessed 17 Dec 2019.
European Medicines Agency. Tailored scientific advice to support step-by-step development of new biosimilars 2018; https://www.ema.europa.eu/en/documents/other/tailored-scientific-advice-support-step-step-development-new-biosimilars_en.pdf. Accessed 17 Dec 2019.
US Food and Drug Administration. Formal meetings between the FDA and sponsors or applicants of BsUFA products: guidance for industry (draft guidance). 2018; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/formal-meetings-between-fda-and-sponsors-or-applicants-bsufa-products-guidance-industry. Accessed 17 Dec 2019.
Calvo B, Martinez-Gorostiaga J, Echevarria E. The surge in biosimilars: considerations for effective pharmacovigilance and EU regulation. Ther Adv Drug Saf. 2018;9:601–8.
Li E, Subramanian J, Anderson S, et al. Development of biosimilars in an era of oncologic drug shortages. Drug Des Devel Ther. 2015;9:3247–55.
European Comission. Rules governing medicinal products in the European Union: Guidelines on pharmacovigilance for medicinal products for human use. 2008; https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-9/pdf/vol9a_09-2008_en.pdf. Accessed 17 Dec 2019.
US Food and Drug Administration. Risk Evaluation and Mitigation Strategies (REMS). 2018; https://www.fda.gov/drugs/drug-safety-and-availability/risk-evaluation-and-mitigation-strategies-rems. Accessed 17 Dec 2019.
US Food and Drug Administration. 21st Century Cures Act. 2018; https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/21stCenturyCuresAct/default.htm. Accessed 17 Dec 2019.
Gottlieb S. Submission to Congress: Food & Drug Administration work plan and proposed funding allocations of FDA innovation account. 2017; https://www.fda.gov/downloads/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/21stCenturyCuresAct/UCM562852.pdf. Accessed 17 Dec 2019.
Smolen JS, Landewe R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76:960–77.
Lau CS, Chia F, Harrison A, et al. APLAR rheumatoid arthritis treatment recommendations. Int J Rheum Dis. 2015;18:685–713.
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken). 2016;68:1–25.
National Institute for Health and Care Excellence. Rheumatoid arthritis in adults: management. 2018; https://www.nice.org.uk/guidance/ng100/resources/rheumatoid-arthritis-in-adults-management-pdf-66141531233989. Accessed 17 Dec 2019.
Deighton C, Hyrich K, Ding T, et al. BSR and BHPR rheumatoid arthritis guidelines on eligibility criteria for the first biological therapy. Rheumatology (Oxford). 2010;49:1197–9.
Cardiel MH, Latin American Rheumatology Associations of the Pan-American League of Associations for R, and Grupo Latinoamericano de Estudio de Artritis R. First Latin American position paper on the pharmacological treatment of rheumatoid arthritis. Rheumatology (Oxford) 2006;45 Suppl 2:ii7–ii22.
Lau CS, Chia F, Dans L, et al. 2018 update of the APLAR recommendations for treatment of rheumatoid arthritis. Int J Rheum Dis. 2019;22:357–75.
National Institute for Health and Care Excellence. Drug treatment for rheumatoid arthritis. 2019; https://pathways.nice.org.uk/pathways/rheumatoid-arthritis#path=view%3A/pathways/rheumatoid-arthritis/drug-treatment-for-rheumatoid-arthritis.xml&content=view-index. Accessed 17 Dec 2019.
Bergstra SA, Branco JC, Vega-Morales D, et al. Inequity in access to bDMARD care and how it influences disease outcomes across countries worldwide: results from the METEOR-registry. Ann Rheum Dis. 2018;77:1413.
Gulacsi L, Brodszky V, Baji P, et al. Biosimilars for the management of rheumatoid arthritis: economic considerations. Expert Rev Clin Immunol. 2015;11(Suppl 1):S43–52.
Kalo Z, Voko Z, Ostor A, et al. Patient access to reimbursed biological disease-modifying antirheumatic drugs in the European region. J Mark Access Health Policy. 2017;5:1345580.
Putrik P, Ramiro S, Kvien TK, et al. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis. 2014;73:198–206.
Baumgart DC, Misery L, Naeyaert S, et al. Biological therapies in immune-mediated inflammatory diseases: can biosimilars reduce access inequities? Front Pharmacol. 2019;10:279.
Putrik P, Ramiro S, Kvien TK, et al. Variations in criteria regulating treatment with reimbursed biologic DMARDs across European countries. Are differences related to country’s wealth? Ann Rheum Dis. 2014;73:2010–21.
Conaghan PG, Hensor EMA, Keenan A-M, et al. Persistently moderate DAS-28 is not benign: loss of function occurs in early RA despite step-up DMARD therapy. Rheumatology. 2010;49:1894–9.
Ceccarelli F, Massafra U, Perricone C, et al. Anti-TNF treatment response in rheumatoid arthritis patients with moderate disease activity: a prospective observational multicentre study (MODERATE). Clin Exp Rheumatol. 2017;35:24–32.
Fan R, Zhong J, Wang ZT, et al. Evaluation of “top-down” treatment of early Crohn’s disease by double balloon enteroscopy. World J Gastroenterol. 2014;20:14479–87.
Hibi T, Panaccione R, Katafuchi M, et al. The 5C concept and 5S principles in inflammatory bowel disease management. J Crohns Colitis. 2017;11:1302–8.
Im JP, Ye BD, Kim YS, et al. Changing treatment paradigms for the management of inflammatory bowel disease. Korean J Intern Med. 2018;33:28–35.
Peyrin-Biroulet L, Sandborn W, Sands BE, et al. Selecting therapeutic targets in inflammatory bowel disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol. 2015;110:1324–38.
Vaughn BP, Shah S, Cheifetz AS. The role of mucosal healing in the treatment of patients with inflammatory bowel disease. Curr Treat Opt Gastroenterol. 2014;12:103–17.
D’Haens GR, Sartor RB, Silverberg MS, et al. Future directions in inflammatory bowel disease management. J Crohns Colitis. 2014;8:726–34.
Peyrin-Biroulet L, Ferrante M, Magro F, et al. Results from the 2nd Scientific Workshop of the ECCO. I: Impact of mucosal healing on the course of inflammatory bowel disease. J Crohns Colitis 2011;5:477–83.
Ordas I, Feagan BG, Sandborn WJ. Early use of immunosuppressives or TNF antagonists for the treatment of Crohn’s disease: time for a change. Gut. 2011;60:1754–63.
Kang B, Choe YH. Early biologic treatment in pediatric Crohn’s disease: catching the therapeutic window of opportunity in early disease by treat-to-target. Pediatr Gastroenterol Hepatol Nutr. 2018;21:1–11.
Sandborn WJ, Hanauer S, Van Assche G, et al. Treating beyond symptoms with a view to improving patient outcomes in inflammatory bowel diseases. J Crohns Colitis. 2014;8:927–35.
Tsui JJ, Huynh HQ. Is top-down therapy a more effective alternative to conventional step-up therapy for Crohn’s disease? Ann Gastroenterol. 2018;31:413–24.
Gomollon F, Dignass A, Annese V, et al. 3rd European evidence-based consensus on the diagnosis and management of Crohn’s disease 2016: part 1: diagnosis and medical management. J Crohns Colitis. 2017;11:3–25.
Harbord M, Eliakim R, Bettenworth D, et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 2: current management. J Crohns Colitis 2017;11:769–84.
Mowat C, Cole A, Windsor A, et al. Guidelines for the management of inflammatory bowel disease in adults. Gut. 2011;60:571–607.
Lichtenstein GR, Loftus EV, Isaacs KL, et al. ACG clinical guideline: management of Crohn’s disease in adults. Am J Gastroenterol. 2018;113:481–517.
Sadowski DC, Bernstein CN, Bitton A, et al. Canadian Association of Gastroenterology clinical practice guidelines: the use of tumour necrosis factor-alpha antagonist therapy in Crohn’s disease. Can J Gastroenterol. 2009;23:185–202.
Bressler B, Marshall JK, Bernstein CN, et al. Clinical practice guidelines for the medical management of nonhospitalized ulcerative colitis: the Toronto consensus. Gastroenterology. 2015;148:1035–1058.e3.
Matsuoka K, Kobayashi T, Ueno F, et al. Evidence-based clinical practice guidelines for inflammatory bowel disease. J Gastroenterol. 2018;53:305–53.
D’Haens GR, Panaccione R, Higgins PD, et al. The London Position Statement of the World Congress of Gastroenterology on Biological Therapy for IBD with the European Crohn’s and Colitis Organization: when to start, when to stop, which drug to choose, and how to predict response? Am J Gastroenterol. 2011;106:199–212.
Allen PB, Peyrin-Biroulet L. Moving towards disease modification in inflammatory bowel disease therapy. Curr Opin Gastroenterol. 2013;29:397–404.
Kang B, Choi SY, Choi YO, et al. Subtherapeutic infliximab trough levels and complete mucosal healing are associated with sustained clinical remission after infliximab cessation in paediatric-onset Crohn’s disease patients treated with combined immunosuppressive therapy. J Crohns Colitis. 2018;12:644–52.
Kang B, Choi SY, Kim HS, et al. Mucosal healing in paediatric patients with moderate-to-severe luminal Crohn’s disease under combined immunosuppression: escalation versus early treatment. J Crohns Colitis. 2016;10:1279–86.
Lee YS, Baek SH, Kim MJ, et al. Efficacy of early infliximab treatment for pediatric Crohn’s disease: a three-year follow-up. Pediatr Gastroenterol Hepatol Nutr. 2012;15:243–9.
Schreiber S, Reinisch W, Colombel JF, et al. Subgroup analysis of the placebo-controlled CHARM trial: increased remission rates through 3 years for adalimumab-treated patients with early Crohn’s disease. J Crohns Colitis. 2013;7:213–21.
Colombel JF, Panaccione R, Bossuyt P, et al. Effect of tight control management on Crohn’s disease (CALM): a multicentre, randomised, controlled phase 3 trial. Lancet. 2018;390:2779–89.
Oh EH, Oh K, Han M, et al. Early anti-TNF/immunomodulator therapy is associated with better long-term clinical outcomes in Asian patients with Crohn’s disease with poor prognostic factors. PLoS One. 2017;12:e0177479.
Rubin DT, Uluscu O, Sederman R. Response to biologic therapy in Crohn’s disease is improved with early treatment: an analysis of health claims data. Inflamm Bowel Dis. 2012;18:2225–31.
Schreiber S, Colombel J-F, Bloomfield R, et al. Increased response and remission rates in short-duration Crohn’s disease with subcutaneous certolizumab pegol: an analysis of PRECiSE 2 randomized maintenance trial data. Am J Gastroenterol. 2010;105:1574–82.
Sandborn WJ, Melmed GY, McGovern DPB, et al. Clinical and demographic characteristics predictive of treatment outcomes for certolizumab pegol in moderate to severe Crohn’s disease: analyses from the 7-year PRECiSE 3 study. Aliment Pharmacol Ther. 2015;42:330–42.
Feagan BG, Lasch K, Lissoos T, et al. Rapid response to vedolizumab therapy in biologic-naive patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2018;17(130–8):e7.
Feagan BG, Rubin DT, Danese S, et al. Efficacy of vedolizumab induction and maintenance therapy in patients with ulcerative colitis, regardless of prior exposure to tumor necrosis factor antagonists. Clin Gastroenterol Hepatol. 2017;15(229–39):e5.
Sands BE, Sandborn WJ, Van Assche G, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease in patients naive to or who have failed tumor necrosis factor antagonist therapy. Inflamm Bowel Dis. 2017;23:97–106.
Paramsothy S, Cleveland NK, Zmeter N, et al. The role of biosimilars in inflammatory bowel disease. Gastroenterol Hepatol. 2016;12:741–51.
Zhao S, Chadwick L, Mysler E, et al. Review of biosimilar trials and data on adalimumab in rheumatoid arthritis. Curr Rheumatol Rep. 2018;20:57.
Rawla P, Sunkara T, Raj JP. Role of biologics and biosimilars in inflammatory bowel disease: current trends and future perspectives. J Inflamm Res. 2018;11:215–26.
Jorgensen KK, Olsen IC, Goll GL, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389:2304–16.
Gabbani T, Deiana S, Annese V. CT-P13: design, development, and place in therapy. Drug Des Devel Ther. 2017;11:1653–61.
Kim NH, Lee JH, Hong SN, et al. Long-term efficacy and safety of CT-P13, a biosimilar of infliximab, in patients with inflammatory bowel disease: a retrospective multicenter study. J Gastroenterol Hepatol. 2019;34:1523–32.
Milassin Á, Fábián A, Molnár T. Switching from infliximab to biosimilar in inflammatory bowel disease: overview of the literature and perspective. Ther Adv Gastroenterol. 2019;12:1756284819842748.
Gonczi L, Gecse KB, Vegh Z, et al. Long-term efficacy, safety, and immunogenicity of biosimilar infliximab after one year in a prospective nationwide cohort. Inflamm Bowel Dis. 2017;23:1908–15.
Kaur P, Chow V, Zhang N, et al. A randomised, single-blind, single-dose, three-arm, parallel-group study in healthy subjects to demonstrate pharmacokinetic equivalence of ABP 501 and adalimumab. Ann Rheum Dis. 2017;76:526–33.
Wynne C, Altendorfer M, Sonderegger I, et al. Bioequivalence, safety and immunogenicity of BI 695501, an adalimumab biosimilar candidate, compared with the reference biologic in a randomized, double-blind, active comparator phase I clinical study (VOLTAIRE®-PK) in healthy subjects. Expert Opin Investig Drugs. 2016;25:1361–70.
Shin D, Lee Y, Kim H, et al. A randomized phase I comparative pharmacokinetic study comparing SB5 with reference adalimumab in healthy volunteers. J Clin Pharm Ther. 2017;42:672–8.
Cohen S, Genovese MC, Choy E, et al. Efficacy and safety of the biosimilar ABP 501 compared with adalimumab in patients with moderate to severe rheumatoid arthritis: a randomised, double-blind, phase III equivalence study. Ann Rheum Dis. 2017;76:1679–87.
Cohen SB, Alonso-Ruiz A, Klimiuk PA, et al. Similar efficacy, safety and immunogenicity of adalimumab biosimilar BI 695501 and Humira reference product in patients with moderately to severely active rheumatoid arthritis: results from the phase III randomised VOLTAIRE-RA equivalence study. Ann Rheum Dis. 2018;77:914–21.
Weinblatt ME, Baranauskaite A, Niebrzydowski J, et al. Phase III randomized study of SB5, an adalimumab biosimilar, versus reference adalimumab in patients with moderate-to-severe rheumatoid arthritis. Arthritis Rheumatol. 2018;70:40–8.
Joensuu JT, Huoponen S, Aaltonen KJ, et al. The cost-effectiveness of biologics for the treatment of rheumatoid arthritis: a systematic review. PLoS One. 2015;10:e0119683.
Ferrario A, Humbert T, Kanavos P, et al. Strategic procurement and international collaboration to improve access to medicines. Bull World Health Organ. 2017;95:720–2.
Severs M, Oldenburg B, van Bodegraven AA, et al. The economic impact of the introduction of biosimilars in inflammatory bowel disease. J Crohns Colitis. 2017;11:289–96.
Gulacsi L, Pentek M, Rencz F, et al. Biosimilars for the management of inflammatory bowel diseases: economic considerations. Curr Med Chem. 2019;26:259–69.
Colombel JF, Ungaro R, Aggarwal S, et al. P692 Efficacy and safety of early biologic treatment of Crohn’s disease in adult and paediatric patients: a systematic review. J Crohns Colitis. 2018;12:S461.
Panaccione R, Colombel JF, Bossuyt P, et al. Long-term cost-effectiveness of tight control for Crohn’s disease with adalimumab-based treatment: economic evaluation beyond 48 weeks of CALM trial. J Crohns Colitis. 2018;1:S074–5.
Pillai N, Dusheiko M, Burnand B, et al. A systematic review of cost-effectiveness studies comparing conventional, biological and surgical interventions for inflammatory bowel disease. PLoS One. 2017;12:e0185500.
Puolakka K, Kautiainen H, Möttönen T, et al. Early suppression of disease activity is essential for maintenance of work capacity in patients with recent-onset rheumatoid arthritis: five-year experience from the FIN-RACo trial. Arthritis Rheum. 2005;52:36–41.
Anis A, Zhang W, Emery P, et al. The effect of etanercept on work productivity in patients with early active rheumatoid arthritis: results from the COMET study. Rheumatology. 2009;48:1283–9.
Cheung WY, Kornelsen EA, Mittmann N, et al. The economic impact of the transition from branded to generic oncology drugs. Curr Oncol. 2019;26:89–93.
Berns M, Hommes DW. Anti-TNF-alpha therapies for the treatment of Crohn’s disease: the past, present and future. Expert Opin Investig Drugs. 2016;25:129–43.
Jha A, Upton A, Dunlop WC, et al. The budget impact of biosimilar infliximab (Remsima®) for the treatment of autoimmune diseases in five European countries. Adv Ther. 2015;32:742–56.
Brodszky V, Baji P, Balogh O, et al. Budget impact analysis of biosimilar infliximab (CT-P13) for the treatment of rheumatoid arthritis in six Central and Eastern European countries. Eur J Health Econ. 2014;15(Suppl 1):S65–71.
Simon Kucher & Partners. Payers’ price & market access policies supporting a sustainable biosimilar medicines market. 2016; https://www.medicinesforeurope.com/wp-content/uploads/2016/09/Simon-Kucher-2016-Policy-requirements-for-a-sustainable-biosimilar-market-FINAL-report_for-publication2.pdf. Accessed 17 Dec 2019.
Khraishi M, Stead D, Lukas M, et al. Biosimilars: a multidisciplinary perspective. Clin Ther. 2016;38:1238–49.
Zheng MK, Shih DQ, Chen GC. Insights on the use of biosimilars in the treatment of inflammatory bowel disease. World J Gastroenterol. 2017;23:1932–43.
Mitrev N, Vande Casteele N, Seow CH, et al. Review article: consensus statements on therapeutic drug monitoring of anti-tumour necrosis factor therapy in inflammatory bowel diseases. Aliment Pharmacol Ther. 2017;46:1037–53.
Papamichael K, Juncadella A, Wong D, et al. Proactive therapeutic drug monitoring of adalimumab is associated with better long-term outcomes compared to standard of care in patients with inflammatory bowel disease. J Crohns Colitis. 2019;13:976–81.
Reinisch W, Eser A, Schreiber S, et al. Body weight and rapid clearance determine early formation of anti-drug antibodies against infliximab. United European Gastroenterol J. 2017;5(suppl 1):A102.
Ricciuto A, Dhaliwal J, Walters TD, et al. Clinical outcomes with therapeutic drug monitoring in inflammatory bowel disease: a systematic review with meta-analysis. J Crohns Colitis. 2018;12:1302–15.
Roblin X, Riviere P, Flamant M, et al. Proactive therapeutic drug monitoring of TNF antagonists in inflammatory bowel disease. Inflamm Bowel Dis. 2018;24:1904–9.
Ben-Horin S. Drug level-based anti-tumor necrosis factor therapy: ready for prime time? Gastroenterology. 2015;148:1268–71.
Martelli L, Olivera P, Roblin X, et al. Cost-effectiveness of drug monitoring of anti-TNF therapy in inflammatory bowel disease and rheumatoid arthritis: a systematic review. J Gastroenterol. 2017;52:19–25.
NHS Improvement. The NHS saves £324 million in a year by switching to better value medicines. 2018; https://improvement.nhs.uk/news-alerts/nhs-saves-324-million-year-switching-better-value-medicines/. Accessed 17 Dec 2019.
Aladul MI, Fitzpatrick RW, Chapman SR. Impact of infliximab and etanercept biosimilars on biological disease-modifying antirheumatic drugs utilisation and NHS budget in the UK. BioDrugs. 2017;31:533–44.
Jahnsen J, Kaasen Jorgensen K. Experience with biosimilar infliximab (Remsima®) in Norway. Dig Dis. 2017;35:83–90.
Lakatos P. European experience of infliximab biosimilars for the treatment of inflammatory bowel disease. Gastroenterol Hepatol. 2016;12:119–21.
Generics and Biosimilars Initiative. France aims to reach 80% biosimilar penetration by 2022. 2018; http://www.gabionline.net/Policies-Legislation/France-aims-to-reach-80-biosimilar-penetration-by-2022. Accessed 17 Dec 2019.
Danese S, Fiorino G, Raine T, et al. ECCO position statement on the use of biosimilars for inflammatory bowel disease-an update. J Crohns Colitis. 2017;11:26–34.
Yoo DH, Prodanovic N, Jaworski J, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76:355–63.
Liu Y, Yang M, Garg V, et al. Economic impact of non-medical switching from originator biologics to biosimilars: a systematic literature review. Adv Ther. 2019;36:1851–77.
Moorkens E, Simoens S, Troein P, et al. Different policy measures and practices between Swedish counties influence market dynamics: part 2-biosimilar and originator etanercept in the outpatient setting. BioDrugs. 2019;33:299–306.
Danese S, Peyrin-Biroulet L. IBD: to switch or not to switch: that is the biosimilar question. Nat Rev Gastroenterol Hepatol. 2017;14:508–9.
Feagan BG, Lam G, Ma C, et al. Systematic review: efficacy and safety of switching patients between reference and biosimilar infliximab. Aliment Pharmacol Ther. 2019;49:31–40.
Blauvelt A, Lacour JP, Fowler JF Jr, et al. Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol. 2018;179:623–31.
Gerdes S, Thaçi D, Griffiths CEM, et al. Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol. 2018;32:420–7.
Kemp K, Dibley L, Chauhan U, et al. Second N-ECCO consensus statements on the European nursing roles in caring for patients with Crohn’s disease or ulcerative colitis. J Crohns Colitis. 2018;12:760–76.
Peyrin-Biroulet L, Lonnfors S, Roblin X, et al. Patient perspectives on biosimilars: a survey by the European Federation of Crohn’s and Ulcerative Colitis Associations. J Crohns Colitis. 2017;11:128–33.
van Overbeeke E, De Beleyr B, de Hoon J, et al. Perception of originator biologics and biosimilars: a survey among Belgian rheumatoid arthritis patients and rheumatologists. BioDrugs. 2017;31:447–59.
Aladul MI, Fitzpatrick RW, Chapman SR. Patients’ understanding and attitudes towards infliximab and etanercept biosimilars: result of a UK web-based survey. BioDrugs. 2017;31:439–46.
Teeple A, Ginsburg S, Howard L, et al. Patient attitudes about non-medical switching to biosimilars: results from an online patient survey in the United States. Curr Med Res Opin. 2019;35:603–9.
Kristensen LE, Alten R, Puig L, et al. Non-pharmacological effects in switching medication: the nocebo effect in switching from originator to biosimilar agent. BioDrugs. 2018;32:397–404.
Wiland P, Batko B, Brzosko M, et al. Biosimilar switching—current state of knowledge. Reumatologia. 2018;56:234–42.
Pouillon L, Socha M, Demore B, et al. The nocebo effect: a clinical challenge in the era of biosimilars. Expert Rev Clin Immunol. 2018;14:739–49.
Scherlinger M, Germain V, Labadie C, et al. Switching from originator infliximab to biosimilar CT-P13 in real-life: the weight of patient acceptance. Joint Bone Spine. 2018;85:561–7.
Boone NW, Liu L, Romberg-Camps MJ, et al. The nocebo effect challenges the non-medical infliximab switch in practice. Eur J Clin Pharmacol. 2018;74:655–61.
Rezk MF, Pieper B. Treatment outcomes with biosimilars: be aware of the nocebo effect. Rheumatol Ther. 2017;4:209–18.
Laugesen J, Hassanein K, Yuan Y. The impact of internet health information on patient compliance: a research model and an empirical study. J Med Internet Res. 2015;17:e143.
Lyman GH, Balaban E, Diaz M, et al. American Society of Clinical Oncology statement: biosimilars in oncology. J Clin Oncol. 2018;36:1260–5.
Cohen H, Beydoun D, Chien D, et al. Awareness, knowledge, and perceptions of biosimilars among specialty physicians. Adv Ther. 2016;33:2160–72.
Jacobs I, Singh E, Sewell KL, et al. Patient attitudes and understanding about biosimilars: an international cross-sectional survey. Patient Prefer Adherence. 2016;10:937–48.
Kim WS, Ogura M, Kwon H-C, et al. Looking to the future and learning lessons from the recent past: changing stakeholder perceptions of biosimilars in cancer. Future Oncol. 2017;13:17–29.
Leonard E, Wascovich M, Oskouei S, et al. Factors affecting health care provider knowledge and acceptance of biosimilar medicines: a systematic review. J Manag Care Spec Pharm. 2019;25:102–12.
Braun J, Kudrin A. Switching to biosimilar infliximab (CT-P13): evidence of clinical safety, effectiveness and impact on public health. Biologicals. 2016;44:257–66.
Danese S, Fiorino G, Michetti P. Changes in biosimilar knowledge among European Crohn’s Colitis Organization [ECCO] members: an updated survey. J Crohns Colitis. 2016;10:1362–5.
Park W, Yoo DH, Miranda P, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76:346–54.
Westhovens R, Yoo DH, Jaworski J, et al. Novel formulation of CT-P13 for subcutaneous administration in patients with rheumatoid arthritis: initial results from a phase I/III randomised controlled trial [THU0191, presented at EULAR 2018]. Ann Rheum Dis. 2018;77:315.
Reinisch W, Jang BI, Borzan V, et al. A novel formulation of CTP13 (infliximab biosimilar) for subcutaneous administration: 1-year result from a phase I open-label randomised controlled trial in patients with active Crohn’s disease [abstract A1103, presented at ECCO 2019].
Schreiber S, Jang BI, Borzan V, et al. Novel formulation of CT-P13 (infliximab biosimilar) for subcutaneous administration: Initial results from a phase I open-label randomized controlled trial in patients with active Crohn’s disease. Gastroenterology. 2018;154:S-1371.
US Food and Drug Administration. Considerations in demonstrating interchangeability with a reference product: guidance for industry. 2019; https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considerations-demonstrating-interchangeability-reference-product-guidance-industry. Accessed 17 Dec 2019.
Acknowledgements
Medical writing support (including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking, and referencing) was provided by Beatrice Tyrrell, DPhil, and Emma Evans, PhD CMPP, at Aspire Scientific (Bollington, UK) and funded by Celltrion Healthcare Co., Ltd (Incheon, Republic of Korea). Celltrion Healthcare Co., Ltd reviewed the article for scientific and medical accuracy. The authors had full editorial control of the article and provided final approval of all content.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
Celltrion Healthcare Co., Ltd provided funding for medical writing support for this article.
Conflicts of interest
HK is employed by and holds stock options in Celltrion Healthcare Co., Ltd. LA has received financial support from Biogen for lectures; grants have been awarded to the European Federation of Crohn’s & Ulcerative Colitis Associations (for projects, advocacy activities, surveys) from Atlantic, AbbVie, Biogen, Celgene, Celltrion, Merck-MSD, Ferring, Janssen, Mundipharma, Otsuka, Pfizer, Shire, Takeda, Vifor, and Philips Morris International Foundation. AD has served as consultant for AbbVie, Celgene, Janssen, MSD, Pfizer, Roche, Sandoz/Hexal, Takeda, and Vifor; received payment for development of educational presentations from Falk Foundation, Ferring, Tillotts, and Takeda; received speaker fees from AbbVie, Falk Foundation, Ferring, Janssen, MSD, Pfizer, Vifor, and Takeda; received manuscript fees from Thieme and Wiley; and received research grants from Institut für Gemeinwohl and Stiftung Leben mit Krebs. FG has received financial support from AbbVie, Janssen, MSD, and Takeda for lectures and travel to scientific meetings. JH has received research grants from Janssen, MSD, and Takeda; served as a consultant and/or an advisory board member for AbbVie, Celgene, Celltrion, Ferring, Hospira, Janssen, Medivir, MSD, Novartis, Pfizer, Prometheus Laboratories Inc., Sandoz, Shire, Takeda, Tillots Pharma, and Vifor Pharma; and served as a speaker for AbbVie, Celgene, Ferring, Hospira, Janssen, MSD, Novartis, Pfizer, Sandoz, Shire, Takeda, Tillotts Pharma, and Vifor Pharma. PMI has received honoraria for acting in an advisory capacity from AbbVie, Celgene, Genentech, Gilead, Hospira, Janssen, Lilly, MSD, Pfizer, Pharmacosmos, Prometheus, Samsung Bioepis, Sandoz, Takeda, Topivert, VH2, Vifor, and Warner Chilcott; received speaker fees from AbbVie, Falk Pharma, Ferring, Janssen, MSD, Pfizer, Sandoz, Shire, Takeda, Tillotts, and Warner Chilcott; and received research funding from MSD, Pfizer, and Takeda. JJ has served as a speaker, consultant, or advisory board member for AbbVie, Astro Pharma, Boehringer Ingelheim, Bristol-Myers Squibb, Celltrion, Ferring, Hikma, Janssen, Meda, MSD, Napp Pharma, Orion Pharma, Pfizer, Pharmacosmos, Roche, Takeda, Tillotts, and Sandoz. PLL has received consulting fees or lecture support from AbbVie, Celgene, Celltrion, Ferring, MSD, Pfizer, Roche, and Takeda and unrestricted research support from AbbVie, Pfizer, and Takeda. BP has received honoraria and speaker fees from AbbVie, AstraZeneca, Celltrion, GSK, Lilly, Pfizer, Roche, and UCB. LP-B has received consulting fees from AbbVie, Allergan, Alma, Amgen, Applied Molecular Transport, Arena, Biogen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Celltrion, Enterome, Ferring, Genentech, Gilead, Index Pharmaceuticals, Janssen, Lilly, MSD, Nestle, Oppilan Pharma, Pfizer, Pharmacosmos, Roche, Samsung Bioepis, Sandoz, Sterna, Sublimity Therapeutics, Takeda, Tillotts, and Vifor; payment for lectures, including service on speakers bureaus, from AbbVie, Biogen, Celltrion, Ferring, Hikma, Janssen, MSD, Pfizer, Roche, Samsung Bioepis, Takeda, and Tillotts; grant/research support from AbbVie, MSD, and Takeda; and stock options from CT-SCOUT. Stefan Schreiber has received personal fees for participation on advisory boards from AbbVie, Arena, Biogen, Bristol-Myers Squibb, Celgene, Celltrion, IMAB, Falk, Fresenius, Gilead, Janssen, MSD, Mylan, Pfizer, Protagonist, Provention Bio, Takeda, and Theravance. Steven Simoens has conducted biosimilar research sponsored by Hospira (now Pfizer); was involved in a stakeholder roundtable on biosimilars sponsored by Amgen, MSD, and Pfizer; has participated in an advisory board meeting for Pfizer; and currently works with Amgen, Celltrion, Mundipharma, and Pfizer as a consultant to carry out biosimilar research. RW has acted as a principal investigator and advisor for Celltrion and Galapagos/Gilead and has given lectures for Celltrion. SD has received consulting fees or honorarium from AbbVie, Allergan, Amgen, AstraZeneca, Biogen, Boehringer Ingelheim, Celgene, Celltrion, Ferring Pharmaceuticals Inc., Gilead, Hospira, Janssen, Johnson & Johnson, MSD, Mundipharma, and Pfizer. RA, KG, JHJ, JL, and SM have no conflicts of interest that are directly relevant to the content of this article.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kim, H., Alten, R., Avedano, L. et al. The Future of Biosimilars: Maximizing Benefits Across Immune-Mediated Inflammatory Diseases. Drugs 80, 99–113 (2020). https://doi.org/10.1007/s40265-020-01256-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01256-5