
Abstract
Despite the widespread use of seasonal influenza vaccines, there is urgent need for a universal influenza vaccine to provide broad, long-term protection. A number of factors underpin this urgency, including threats posed by zoonotic and pandemic influenza A viruses, suboptimal effectiveness of seasonal influenza vaccines, and concerns surrounding the effects of annual vaccination. In this article, we discuss approaches that are being investigated to increase influenza vaccine breadth, which are near-term, readily achievable approaches to increase the range of strains recognized within a subtype, or longer-term more challenging approaches to produce a truly universal influenza vaccine. Adjuvanted and neuraminidase-optimized vaccines are emerging as the most feasible and promising approaches to extend protection to cover a broader range of strains within a subtype. The goal of developing a universal vaccine has also been advanced with the design of immunogenic influenza HA-stem constructs that induce broadly neutralizing antibodies. However, these constructs are not yet sufficiently immunogenic to induce lasting universal immunity in humans. Advances in understanding how T cells mediate protection, and how viruses are packaged, have facilitated the rationale design and delivery of replication-incompetent virus vaccines that induce broad protection mediated by lung-resident memory T cells. While the lack of clear mechanistic correlates of protection, other than haemagglutination-inhibiting antibodies, remains an impediment to further advancing novel influenza vaccines, the pressing need for such a vaccine is supporting development of highly innovative and effective strategies.
Similar content being viewed by others

Current influenza vaccines provide only moderate protection that is largely strain-specific. |
The development of influenza vaccines with increased breath and efficacy is an achievable short-term goal that should be pursued. |
Universal influenza vaccine development represents a considerable challenge that relies on inducing rare B cells that make fully cross-protective antibodies, and/or T cells that can be retained in sufficient numbers in the mucosa. |
1 Introduction
Influenza viruses comprise four types, A, B, C and D, of which types A, B and C infect humans and types A and B cause influenza epidemics. Influenza A viruses are subtyped based on the antigenicity of the envelope haemagglutinin (HA) and neuraminidase (NA) proteins into 18 HA and 11 NA subtypes, which occur in a wide range of animal species, including waterfowl and shorebirds, pigs, horses and dogs. The HA is a trimer, with a globular head that bears the receptor-binding pocket and neutralizing antibody-binding sites, and a stem (or stalk) that includes a peptide that mediates fusion of virus and host membranes during virus entry [1]. The 18 influenza A virus HAs are classified into groups 1 and 2 based on phylogenetic similarity of the stem region. Influenza viruses that currently circulate in humans include two influenza A subtypes, H1N1 (group 1) and H3N2 (group 2), and two influenza B lineages, Yamagata and Victoria. Influenza viruses mutate readily because the RNA-dependent RNA polymerase lacks proof-reading function, and selection pressure leads to the rapid generation of viruses containing mutations in antibody binding sites. This process, termed antigenic drift, is ongoing, difficult to predict and necessitates frequent updates of the influenza vaccine. The establishment of an influenza virus with a novel HA subtype to which the majority of the population lacks immunity is referred to as antigenic shift. Other avian influenza A subtypes, including H5N1, H7N9, H6N1, and H7N7, have infected humans but have not transmitted efficiently between humans, whereas H2N2 virus caused a pandemic in 1957 and caused epidemic influenza in humans until 1968.
Seasonal trivalent or quadrivalent influenza vaccines, including inactivated influenza vaccine (IIV), live attenuated influenza vaccine (LAIV), and recombinant HA vaccines, include antigens from each of the type A and B viruses that cause epidemic influenza. They induce strain-specific antibody-mediated immunity to the HA, with limited effectiveness against antigenic drift variants within the same subtype and virtually none against other influenza A virus subtypes. Ongoing antigenic drift in seasonal influenza A and B viruses, suboptimal effectiveness of currently licensed influenza vaccines [2] that can be of very limited duration in the elderly [3], and the sporadic threats of zoonotic and pandemic influenza A viruses are driving efforts to develop broadly protective influenza vaccines. This goal encompasses a spectrum that extends from a near-term achievable goal of a vaccine with enhanced efficacy against antigenic drift variants within the same subtype, to a long-term, more distant goal of a truly universal influenza vaccine that protects against all influenza A and B viruses (Fig. 1). Different strategies along this spectrum will provide increasing breadth of coverage against antigenic drift variants, zoonotic strains and pandemic influenza viruses.

Comparison of strategies to increase the breadth of influenza vaccines in terms of the anticipated duration of the immune responses invoked and timescales for product development. IIV inactivated influenza vaccine, HA hemagglutinin, NA neuraminidase
2 Strategies to Induce a More Broadly-Reactive Immune Response
2.1 New Approaches for Vaccine Strain Selection
The WHO Global Influenza Surveillance and Response System (GISRS) has operated since the 1950s to select seasonal influenza vaccine strains that closely match circulating strains, genetically and antigenically. GISRS also characterizes zoonotic influenza viruses to update candidate vaccine viruses for pandemic preparedness (http://www.who.int/influenza/vaccines/virus/201602_zoonotic_vaccinevirusupdate.pdf). It is not known which animal influenza virus may emerge to cause a pandemic. Therefore, the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH)/MedImmune collaborative programme undertook identification of animal influenza viruses that would generate antibodies with broad cross-reactivity against viruses within the same subtype. Ferret anti-sera were generated against 10–20 viruses each for H2 [4], avian and equine H3 [5], H5 [6], H6 [7, 8], H7 [9] and H10 [10] subtypes, including viruses that covered a wide geographic and temporal distribution and that represented different phylogenetic clades. Virus(es) that induced the greatest breadth of cross-reactivity in haemagglutination-inhibiting (HI) and neutralization assays were used to generate candidate pandemic LAIVs, which were evaluated in animal models and in phase I clinical trials [4, 11,12,13,14,15,16]. An alternative approach has been to design a computationally optimized broadly reactive antigen (COBRA) based on multiple rounds of generating HA consensus sequences. COBRA-HA virus-like particle (VLP) vaccines incorporating H5-HA or H1-HA induce cross-clade antibodies in mice [17, 18].
Several groups have explored forecasting of influenza A(H3N2) and A(H1N1) virus antigenic evolution by incubating viruses with human and/or ferret convalescent sera and driving positive selection [19, 20]. This approach suggests that it may be possible to make vaccines containing future antigenic escape variants and pre-empt antigenic drift.
2.2 Adjuvants
Adjuvants augment the adaptive immune response to vaccines, either by triggering innate immunity through pathogen recognition receptors (PRRs) with pathogen- or damage-associated molecular patterns (PAMPs and DAMPs) or by facilitating antigen delivery. Humoral immunity following influenza virus infection is facilitated by a broad array of PRRs, TLR7, TLR3, RIG-I/MAVS and the NLRP3 inflammasome (reviewed by Iwasaki and Pillai [21]). In contrast, the standard subunit and split virion IIV are unadjuvanted, and immunogenicity is more reliant on activation of TLR7, which recognizes single-stranded viral RNA [22].
The modest immunogenicity of IIV for high-risk groups has led to the clinical development of adjuvants. MF59, a squalene-based oil-in-water emulsion, was approved by the US FDA in 2015 for use in the elderly. Compared with IIV, adjuvanting with MF59 has been shown to increase antibody titre and cross-protection in older individuals [23, 24] and children [25]. For example, among elderly vaccinees, MF59-adjuvanted vaccine generates protective levels of antibody in 98%, whereas unadjuvanted vaccine generates this in 70–80% [23]. AS03, developed by GlaxoSmithKline, is another oil-in-water emulsion that uses squalene and dl-α-tocopherol. AS03 has been approved for several different pandemic influenza vaccine formulations, including Pandemrix™ used in Europe during the 2009 pandemic, Adjupanrix™ for A/H5N1 in Europe, and Q-Pan for A/H5N1 in the US. In general, AS03 induces robust antibody responses, with observations of 100% seroconversion against homologous strains and 69% seroconversion against heterologous strains in clinical trials [26, 27]. A head-to-head comparison showed that H7N9 vaccine titres were far higher when adjuvanted with AS03 compared with MF59 [28]. ISCOMATRIX, a formulation comprised of saponin, phospholipid and cholesterol, also increases seroconversion rates against an H7N9 VLP vaccine [29, 30]. Antibodies induced by unadjuvanted and ISCOMATRIX-adjuvanted VLPs were compared for binding to an HA gene fragment phage display library [30]. Antibodies induced by unadjuvanted VLPs predominantly bound epitopes in the C-terminus of the HA1 domain, whereas antibodies induced by ISCOMATRIX-adjuvanted VLPs also bound more accessible, conformational epitopes spanning the receptor binding domain, and are therefore more likely to mediate HI and protection [30].
AS03 and MF59 augment humoral immunity by increasing naive B-cell activation, B-cell receptor (BCR) adaptation by recalled memory B cells [31], and antibody persistence in ferrets [32] and humans [33]. It is assumed that they facilitate antigen uptake, but the actual mechanisms of adjuvancy are not known. Neither include PAMPs, so there remains scope to combine PAMPs, DAMPs and other antigen delivery modalities to promote robust adaptive immunity. Indeed, several experimental adjuvants using PAMPs and DAMPs show promise. TLR5 sensing is critical for optimal antibody responses to IIV, even though IIV does not directly engage TLR5 [34]. It is thought that flagellin, produced by gut microbiota, triggers TLR5-mediated priming of innate mechanisms that promote plasma cell differentiation [34]. Fusion proteins of influenza antigens and flagellin are being clinically tested [35, 36]. S-[2,3-bis(palmitoyloxy)propyl]-cysteine or Pam2Cys and R4Pam2Cys are TLR2 agonists that increase the immunogenicity of influenza-derived peptides and detergent-split IIV, respectively, leading to increased protection against homologous and heterologous challenge in mice [37,38,39]. R4Pam2Cys associates electrostatically with oppositely charged regions on protein antigens, forming antigen complexes that can be directed to TLR2 on dendritic cells, and induce both CD8+ T-cell and antibody responses [38, 40].
The inclusion of adjuvants can lead to higher rates of vaccine-associated side effects. These are generally very mild, such as inflammation at the injection site, fever and headache, but very rarely they can be more pronounced and serious. For example, small but significant increases in the rates of narcolepsy were observed in recipients of the Pandemrix™ vaccine in a number of countries [41, 42]. While there was initial concern that the AS03 adjuvant triggered narcolepsy, the adjuvant has since been used in a number of other vaccine formulations without similar observations. The precise cause of narcolepsy with Pandemrix™ remains unknown but is being extensively examined.
As a possible alternative to adjuvants, antibodies have been used to form antigen immune complexes that can engage Fcγ receptors on B cells [43]. Stimulation of Fcγ receptors, such as CD23, is dependent on the level of immunoglobulin (Ig)G-Fc glycosylation [44]. Interestingly, levels of Fc-glycosylated anti-HA IgG peak approximately 1 week after vaccination with the trivalent IIV, and complexing with Fc-glycosylated anti-HA IgG induces more potent and broadly reactive antibodies [45]. Fc-glycosylated IgG facilitates affinity selection by engaging CD23 on antigen-specific B cells; this induces FcγRIIB and inhibits the maturation of low but not high affinity B cells [45]. IIV complexed with a broadly HA-reactive mAb that engages CD23 protects mice against influenza H5N1 virus challenge [46].
As outlined above, there is both scope for, and a need for, improved adjuvants in influenza vaccines, and this will continue to drive development and refinement of formulations into the future.
2.3 Neuraminidase-Inhibiting Antibodies
The viral NA cleaves sialic acids from the surface of infected cells, releasing progeny virions [47, 48]. Therefore, although NA-inhibiting (NI) antibodies do not block virus entry or neutralize infectivity [47], they reduce virus release and spread, and in turn reduce illness severity in animal models [49, 50]. The evidence for a protective role of NI antibodies in humans is strong. When H3N2 viruses emerged in the 1968 pandemic, replacing previously circulating H2N2 viruses, H3-specific HI antibodies were lacking but partial protection associated with N2-reactive NI antibodies was demonstrated [51,52,53]. Clinical trials conducted in the 1970s and 1980s also demonstrated that NA-only vaccines protected children [54] and adults [55] against influenza illness, while being permissive to infection. Since then, more reliable, higher throughput NI assays such as the enzyme-linked lectin assay (ELLA) have been developed [56, 57] and several studies have correlated NI titres with protection against illness [58,59,60,61]. Antigenic drift of the HA and NA are not coordinated, therefore NI antibodies may provide protection against HA drift variants [62]. More importantly, NI antibodies provide cross-strain protection in animal models [63,64,65], consistent with the presence of conserved epitopes in N1 and N2 that are recognized by NI antibodies from mice [66] and humans [67]. A striking observation has been that while antibodies with broad NI activity are induced in humans following influenza infection, they are not induced following vaccination, suggesting that the NA content or structural integrity of NA in IIV is suboptimal [67]. A potential ‘universal NA’-inhibiting antibody epitope has also been identified [68].
While the combined evidence supports the development of NA-optimized vaccines, the NA concentration is not standardized in current vaccines; it varies widely and tends to be low [69]. To optimize NA concentration, it is important to first define the optimal protective NI titre, using assays such as ELLA. It may also be necessary to use NA-only vaccines to define protective NI titres and overcome the immunodominance of the HA head [70,71,72], with the potential benefit that NA-only vaccines should prevent symptoms but permit natural infection that will boost a broader range of immune responses than conventional IIV [73, 74].
2.4 Haemagglutinin Stem-Based Vaccines
HA head-directed neutralizing antibodies rarely recognize conserved regions such as the receptor binding site (reviewed by Neu et al. [75]). The highly conserved HA stem elicits protective antibodies that neutralize virus infectivity without inhibiting haemagglutination, and is a more attractive target for the development of a universal influenza vaccine. The HA stem also induces non-neutralizing antibodies that inhibit virus infection by mechanisms such as antibody-dependent cell-mediated cytotoxicity (ADCC). The ability of both neutralizing and non-neutralizing HA stem antibodies to protect humans against influenza has not yet been documented. Moreover, the two categories of HA stem antibodies may not be equivalent in their protective efficacy. Four epitopes have been identified in the stem region of group 1 and 2 influenza A and B viruses (reviewed by Neu et al. [75]). Although escape mutations arise readily in the HA head, the stem epitope is not normally under immune pressure, and attempts to generate escape mutants in vitro suggest that they will not arise easily [76].
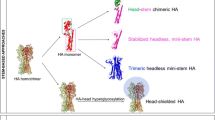
The biggest technical challenge in designing an HA stem vaccine is to direct the antibody response towards the stem and away from the immunodominant HA head. The first approach that was attempted was a ‘headless’ construct [77], but a portion of the stem epitope that is part of the HA1 domain was missing from this construct. Subsequently, a strategy was developed for serial immunization with chimeric HA molecules that were engineered with different head domains atop an identical stem (reviewed by Neu et al. [75]). The underlying principle is that the antibody response to each new head domain will be a primary response, while the response to the stem will be boosted because it is identical in all the chimeric constructs [78]. The plan is to administer prime-boost vaccinations with chimeric HAs as LAIV and IIV, respectively. This approach has shown great promise in animal models and is now in a phase 1 clinical trial.
Polypeptides mimicking the native, pre-fusion HA stem epitope of group 1 influenza A viruses have also shown very promising results in animal models [79,80,81,82]. Yassine et al. used iterative cycles of a structure-based design to develop an H1 HA stabilized-stem immunogen lacking the immunodominant head domain, presented on a ferritin nanoparticle [83]. This immunogen conferred complete heterosubtypic protection against lethal H5N1 challenge in mice and partial protection in ferrets (4/6 survived) by eliciting broadly cross-reactive antibodies. However, the development of a group 2 HA stem vaccine has been more challenging, likely due to structural and/or biochemical differences between group 1 and group 2 HAs [84]. In collaboration with scientists from the Indian Institute of Science, we evaluated a bacterially expressed group 2 stem immunogen that was confirmed by biophysical characterization to form folded trimeric proteins. Vaccination induced stem-directed antibodies that protected mice from lethal homologous and intrasubtypic challenge and provided moderate protection against lethal heterologous virus challenge. However, in ferrets, vaccination induced relatively low levels of HA stem-directed antibodies that did not significantly reduce weight loss or nasal wash titres following robust H7N9 virus challenge. Epitope mapping revealed that ferrets developed lower titres of antibodies that bound a narrower range of HA stem epitopes than mice. We infer that this likely explains the lower efficacy in ferrets [84]. Our findings indicate that while group 2 stem immunogen showed promise in inbred mice, their immunogenicity and efficacy in larger outbred animals was not yet optimal and needs to be enhanced.
Once the technical challenge of inducing a robust stem-specific immune response is solved, downstream challenges include demonstrating efficacy against novel viruses in humans, or identifying correlate(s) of protection, characterizing the longevity of the response and developing appropriate assays to quantify them.
2.5 Vaccines to Induce Influenza-Reactive T Cells
T cells recognize internal proteins of the virus that are conserved and are not accessible to antibodies, have the capacity to kill virus-infected cells in vitro, and contribute to influenza virus clearance and control of infection in mouse models [85, 86]. Therefore, vaccines that induce robust T-cell responses have the potential to provide protection against influenza viruses bearing novel envelope proteins. IIVs induce little, if any, T-cell response. LAIV induces influenza-reactive T cells in animal models [87,88,89] and humans [90,91,92,93], and may induce greater heterologous immunity than IIV, at least in young children [94,95,96], but induces little or no heterosubtypic immunity [97]. The US Advisory Committee on Immunization Practices recommended against the use of LAIV for two recent seasons due to low effectiveness against the A(H1N1)pdm09 virus, which was postulated to reflect decreased replicative fitness [98]. A fitter A(H1N1)pdm09 strain has been included in 2018–2019 LAIV, and the recommendation against LAIV has been removed [98].
Depletion or functional impairment of individual CD4+ or CD8+ T-cell subsets does not uniformly exacerbate disease, indicating that multiple mechanisms contribute to heterosubtypic immunity [99, 100]. Some studies indicate that the contribution of CD4+ T cells is limited in the absence of CD8+ T and B cells [101], and that CD8+ T cells are the effectors of virus clearance [102, 103]. The requirement for more control mechanisms as the virus dose increases [104] could account for discrepant findings regarding the need for CD4+ [105] versus CD8+ T cells [106, 107] for protection against influenza illness in humans.
CD4+ T cells play a central role in the generation of high-affinity, isotype-switched B cells and antibodies that provide long-term protection against re-infection. Affinity selection occurs within germinal centres and is mediated by CD4+ T follicular helper (Tfh) cells that characteristically express the chemokine receptor CXCR5 to facilitate entry into lymphoid follicles [108, 109]. Whether changes in circulating Tfh [108, 110] can inform vaccine development [111] is an area of active investigation. Influenza vaccination causes transient expansion of peripheral CXCR3+, ICOS+, CD38+ Tfh cells, which include influenza-specific cells [111, 112]. Limited studies in mice indicate that optimal HI antibody responses require HA-specific Tfh [110]. This contrasts with reports that healthy human CD4+ T-cell responses focus on the internal viral nucleoprotein (NP) and matrix (M) proteins [113]. Vaccine-induced Tfh expansion is positively associated with antibody production [111] and benefits of high-dose vaccine in the elderly [114]. Therefore, CD4+ T-cell specificity or Tfh magnitude should be considered for the development of more effective vaccines.
Accumulating evidence indicates that influenza-reactive T cells must reside in the respiratory mucosa for timely control of viral replication and ensuing illness since it takes 4–5 days for memory T cells to track from lymph nodes to the respiratory tract, although they produce interferon (IFN)-γ within 6 h of infection [86]. Additionally, delayed T-cell responses, combined with large amounts of virus in lungs, could contribute to immunopathology [115]. It has been established that tissue resident memory (Trm) T cells represent a distinct subset [116,117,118,119,120,121] that co-express CD69 and CD103, unlike circulating T cells [120, 122,123,124]. The induction of Trm following pulmonary, but not systemic, immunization coincides with reduced viral replication and lung damage [121], and increased protection of mice against lethal influenza virus challenge [125]. Similarly, CD4+ and influenza-reactive CD8+ Trm are induced in mice by LAIV, but not IIV, and mediate heterosubtypic protection independent of circulating T cells and antibody [126].
The susceptibility of humans to repeated influenza infections indicates that T-cell-mediated heterosubtypic immunity may be weak or poorly maintained. A few studies have examined influenza-reactive T-cell frequencies over time in small sample sizes. The frequency of influenza-reactive cells with lytic capacity declines rapidly after infection, with an approximate half-life of 2–3 years [127], but tetramer staining indicates that CD8+ memory T-cell frequencies remain stable in the absence of documented re-infection [128]. Studies to determine whether vaccines induce and maintain Trm will not be possible in humans, but the evidence from preclinical models suggests that T-cell-based vaccines should be delivered mucosally.
Plasmid-based reverse genetics is being used to rationally design influenza vaccines that can infect airway cells to induce Trm, but not replicate because a crucial gene has been rendered defective [129]. For example S-FLU can only undergo a single cycle of replication because the HA packaging signal sequence has been inactivated [129]. HA is provided in trans during pseudotype particle production in cell lines, therefore S-FLU virus can infect cells and express the NA and conserved internal viral proteins in the cytosol, but cannot replicate or donate its HA to other influenza strains. Other single-cycle vaccines have also been produced, most of which lack fully functional HA [130,131,132] or M2 alone [133, 134], whereas Si et al. target multiple gene segments by introducing premature stop codons that can only be translated using a packaging cell line that expresses the appropriate non-host tRNA–tRNA synthetase pair [135]. Similarly, codon-pair deoptimization of specific gene segments yields influenza virus that is highly attenuated for growth, but not immunogenicity, in mice [136], although the optimal balance between attenuation and immunogenicity for other species requires fine tuning [137]. Single replication cycle influenza viruses represent a safer alternative to LAIV that can be delivered intranasally to high-risk and immunocompromised patients [135, 138], with potential to stimulate immune responses in the lower airways. S-FLU vaccines induce a strong cross-reactive T-cell response in the lungs, a specific antibody response to the expressed NA, but a minimal antibody response to the HA pseudotyping the particle [88, 139]. H1N1 or H5N1 S-FLU vaccines induce robust protection against the homologous and heterologous H1N1, H6N1 (group 1), H3N2 and H7N9 (group 2) viruses in mice [88, 139]. Importantly, ferrets immunized with one dose of H1 S-FLU then challenged with the homologous H1N1pdm09 virus did not transmit challenge virus to naive ferrets by the airborne route [88]. In pigs, immunization reduced the viral load in nasal swabs and lungs following challenge with a swine H1N1pdm09 isolate [139]. The ability of aerosol-administered H3N2 S-FLU vaccine to protect against heterosubtypic H1N1pdm09 challenge has been evaluated in pigs and ferrets [140]. H3N2 S-FLU reduced heterosubtypic challenge viral replication and aerosol transmission in ferrets, and induced lung Trm cells and reduced lung pathology, but not the viral load in the upper respiratory tract samples collected daily or bronchoalveolar lavage collected on day 5 at necropsy in pigs. Taken together, S-FLU vaccines showed protective efficacy in pigs and ferrets, demonstrating that in the absence of antibody, lung T-cell immunity can reduce disease severity and lung pathology, even without reducing challenge viral replication in the upper respiratory tract.
Vaccines based on peptide, DNA or viral vectors may induce more robust T-cell responses (discussed below), but further development of T-cell-based vaccines is hindered by a lack of a correlate of protection, and of strategies to induce and maintain influenza-reactive T cells in the respiratory tract. The minimal magnitude, breadth and functional capacity of T cells required for protection need to be defined [105,106,107].
2.5.1 Peptide-Based T-Cell Vaccines
Synthetic peptides are relatively safe and can be readily modified in the event that mutations are identified. However, the peptide vaccine approach requires peptides that are recognized by a diverse spectrum of HLA A and B types in the population. The majority of influenza-specific CD8+ T cells recognize NP and M proteins [141,142,143,144]. Importantly, NP and M contain epitopes presented by common HLAs such as A2, and responses are boosted by infection with different strains [143, 145, 146], reflecting the presence of epitopes that are highly conserved across the majority of influenza A viruses characterized [147, 148]. However, the focus on common HLA types means that immunogenic peptides identified to date do not cover the entire population. For example, the coverage of immunogenic NP peptides is approximately 16–57%, with an absence of epitopes for HLA-A*0101, A*6801, B*1501 and A*2402 [149].
Two influenza peptide vaccines have been assessed in phase I and II clinical trials, and both induce T-cell responses. Flu-v contains 21–33 amino acid stretches of M1, M2 and NP [150, 151], and includes five known CD8+ T-cell epitopes [149]. M-001 contains five T-cell and four B-cell linear peptide epitopes from NP, M1 and HA. Participants immunized twice with M-001 followed by trivalent IIV had significantly elevated T-cell responses [152], seroconversion rates, and antibody titres against drifted strains [153] compared with those who received trivalent IIV alone. While it is assumed that a combination of immune responses may be desirable for optimal protection, validation is required. Clinical trials that examined adjuvant effects [150] indicate the need to target peptides to professional antigen-presenting cells for optimal T-cell activation [154].
2.5.2 Vectored Vaccines
Replication-deficient viral vectors such as modified Vaccinia Ankara (MVA) can be engineered to express multiple protein antigens intracellularly, and thereby induce CD8+ T-cell responses without the need to identify epitopes for particular HLA types, or to use adjuvants [155]. The development of immunity to the vector can limit the use of the same viral vector for repeated (prime and boost) vaccine doses [155]. Therefore, DNA or distinct viral vectors are often used for priming. MVA expressing influenza NP + M1 induces T-cell responses and reduces clinical illness in human challenge studies [156], and can be coadministered with IIV without compromising antibody responses [157]. Similarly, viral vectors incorporating HA, NP and M1 induce robust T-cell responses, long-lived antibody secreting cells and HA-reactive neutralizing antibodies in mice [158]. However, in macaques, two doses of MVA-HA induced potent serum antibody responses against viruses with homologous HAs, but did not stimulate strong T-cell responses prior to challenge [159]. Although post-challenge CD4+ and CD8+ T-cell boosting was observed in animals that received either MVA-HA or MVA-NP, only MVA-HA reduced challenge virus replication and provided protection [159].
2.6 Combining Vaccine Platforms
The combination of different vaccine platforms has been demonstrated to increase the magnitude and breadth of the immune response. In phase I clinical trials evaluating H5N1 vaccine candidates, priming with LAIV [160, 161], or DNA expressing the influenza H5 HA, or adenovirus-H5 HA followed by IIV boosting was more immunogenic than priming and boosting with IIV [160, 162]. Similar results have been obtained from studies combining H7N7 and H7N9 vaccine candidate formulations [16, 163], and, in all studies, immunogenicity was increased by extending the interval between prime and boost [162, 163]. The mechanism underlying the superior priming effects of LAIV over IIV were explored in a non-human primate model [164]. Intranasal H5N1 LAIV elicits a highly localized germinal centre B-cell response in the mediastinal lymph node, which is rapidly recalled following IIV boost, eliciting germinal centre reactions at numerous distant immune sites [164]. These data provide mechanistic insights for the generation of robust humoral responses via prime-boost vaccination.
3 Conclusions
A number of avenues are being pursued to develop broadly protective influenza vaccines against seasonal influenza and, importantly, viruses with pandemic potential. Key drivers are the high rate of enzootic H5Nx and H7N9 viruses in poultry, with hundreds of spillover infections in humans, and the challenges in implementing a monovalent H1N1pdm09 vaccine following the 2009 H1N1 pandemic, in a timely fashion. Clearly, it is possible to increase the breadth of protection conferred by conventional vaccines through a number of approaches, such as the use of adjuvants or viral vectors, the addition of NA, vaccines containing computationally optimized or antigenically advanced HAs, and the selection of strains for inclusion in the vaccine that elicit a more broadly reactive immune response than conventional vaccines, including naturally occurring viruses. New vaccines that target conserved epitopes on the HA stem, or induce cellular immunity against internal viral proteins, induce greater breadth of reactivity and protection than conventional influenza vaccines and show great promise in animal models. Hopefully one or more of these avenues will prove to be safe and effective in humans.
References
Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, et al. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol. 2009;16(3):265–73.
Belongia EA, Simpson MD, King JP, Sundaram ME, Kelley NS, Osterholm MT, et al. Variable influenza vaccine effectiveness by subtype: a systematic review and meta-analysis of test-negative design studies. Lancet Infect Dis. 2016;16(8):942–51.
Young B, Zhao X, Cook AR, Parry CM, Wilder-Smith A, I-Cheng IC. Do antibody responses to the influenza vaccine persist year-round in the elderly? A systematic review and meta-analysis. Vaccine. 2017;35(2):212–21.
Chen GL, Lamirande EW, Jin H, Kemble G, Subbarao K. Safety, immunogencity, and efficacy of a cold-adapted A/Ann Arbor/6/60 (H2N2) vaccine in mice and ferrets. Virology. 2010;398(1):109–14.
Baz M, Paskel M, Matsuoka Y, Zengel J, Cheng X, Jin H, et al. Replication and immunogenicity of swine, equine, and avian h3 subtype influenza viruses in mice and ferrets. J Virol. 2013;87(12):6901–10.
Boonnak K, Matsuoka Y, Wang W, Suguitan AL Jr, Chen Z, Paskel M, et al. Development of clade-specific and broadly reactive live attenuated influenza virus vaccines against rapidly evolving H5 subtype viruses. J Virol. 2017;91(15):1–13.
Gillim-Ross L, Santos C, Chen Z, Aspelund A, Yang CF, Ye D, et al. Avian influenza h6 viruses productively infect and cause illness in mice and ferrets. J Virol. 2008;82(21):10854–63.
Chen Z, Santos C, Aspelund A, Gillim-Ross L, Jin H, Kemble G, et al. Evaluation of live attenuated influenza a virus h6 vaccines in mice and ferrets. J Virol. 2009;83(1):65–72.
Joseph T, McAuliffe J, Lu B, Jin H, Kemble G, Subbarao K. Evaluation of replication and pathogenicity of avian influenza a H7 subtype viruses in a mouse model. J Virol. 2007;81(19):10558–66.
Sutton TC, Lamirande EW, Czako R, Subbarao K. Evaluation of the biological properties and cross-reactive antibody response to H10 influenza viruses in ferrets. J Virol. 2017;91(19):1–18.
Joseph T, McAuliffe J, Lu B, Vogel L, Swayne D, Jin H, et al. A live attenuated cold-adapted influenza A H7N3 virus vaccine provides protection against homologous and heterologous H7 viruses in mice and ferrets. Virology. 2008;378(1):123–32.
Talaat KR, Karron RA, Callahan KA, Luke CJ, DiLorenzo SC, Chen GL, et al. A live attenuated H7N3 influenza virus vaccine is well tolerated and immunogenic in a Phase I trial in healthy adults. Vaccine. 2009;27(28):3744–53.
Min JY, Vogel L, Matsuoka Y, Lu B, Swayne D, Jin H, et al. A live attenuated H7N7 candidate vaccine virus induces neutralizing antibody that confers protection from challenge in mice, ferrets, and monkeys. J Virol. 2010;84(22):11950–60.
Talaat KR, Karron RA, Luke CJ, Thumar B, McMahon BA, Chen GL, et al. An open label Phase I trial of a live attenuated H6N1 influenza virus vaccine in healthy adults. Vaccine. 2011;29(17):3144–8.
Young N, Pebody R, Smith G, Olowokure B, Shankar G, Hoschler K, et al. International flight-related transmission of pandemic influenza A(H1N1)pdm09: an historical cohort study of the first identified cases in the United Kingdom. Influenza Other Respir Viruses. 2014;8(1):66–73.
Babu TM, Levine M, Fitzgerald T, Luke C, Sangster MY, Jin H, et al. Live attenuated H7N7 influenza vaccine primes for a vigorous antibody response to inactivated H7N7 influenza vaccine. Vaccine. 2014;32(50):6798–804.
Giles BM, Ross TM. Computationally optimized antigens to overcome influenza viral diversity. Expert Rev Vaccines. 2012;11(3):267–9.
Carter DM, Darby CA, Lefoley BC, Crevar CJ, Alefantis T, Oomen R, et al. Design and characterization of a computationally optimized broadly reactive hemagglutinin vaccine for H1N1 influenza viruses. J Virol. 2016;90(9):4720–34.
DeDiego ML, Anderson CS, Yang H, Holden-Wiltse J, Fitzgerald T, Treanor JJ, et al. Directed selection of influenza virus produces antigenic variants that match circulating human virus isolates and escape from vaccine-mediated immune protection. Immunology. 2016;148(2):160–73.
Li C, Hatta M, Burke DF, Ping J, Zhang Y, Ozawa M, et al. Selection of antigenically advanced variants of seasonal influenza viruses. Nat Microbiol. 2016;1(6):16058.
Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14(5):315–28.
Geeraedts F, Goutagny N, Hornung V, Severa M, de Haan A, Pool J, et al. Superior immunogenicity of inactivated whole virus H5N1 influenza vaccine is primarily controlled by Toll-like receptor signalling. PLoS Pathog. 2008;4(8):1–8.
Del Giudice G, Hilbert AK, Bugarini R, Minutello A, Popova O, Toneatto D, et al. An MF59-adjuvanted inactivated influenza vaccine containing A/Panama/1999 (H3N2) induced broader serological protection against heterovariant influenza virus strain A/Fujian/2002 than a subunit and a split influenza vaccine. Vaccine. 2006;24(16):3063–5.
Ansaldi F, Bacilieri S, Durando P, Sticchi L, Valle L, Montomoli E, et al. Cross-protection by MF59-adjuvanted influenza vaccine: neutralizing and haemagglutination-inhibiting antibody activity against A(H3N2) drifted influenza viruses. Vaccine. 2008;26(12):1525–9.
Vesikari T, Groth N, Karvonen A, Borkowski A, Pellegrini M. MF59-adjuvanted influenza vaccine (FLUAD) in children: safety and immunogenicity following a second year seasonal vaccination. Vaccine. 2009;27(45):6291–5.
Langley JM, Frenette L, Ferguson L, Riff D, Sheldon E, Risi G, et al. Safety and cross-reactive immunogenicity of candidate AS03-adjuvanted prepandemic H5N1 influenza vaccines: a randomized controlled phase 1/2 trial in adults. J Infect Dis. 2010;201(11):1644–53.
Izurieta P, Kim WJ, Wie SH, Lee J, Lee JS, Drame M, et al. Immunogenicity and safety of an AS03-adjuvanted H5N1 pandemic influenza vaccine in Korean adults: a phase IV, randomized, open-label, controlled study. Vaccine. 2015;33(24):2800–7.
Jackson LA, Campbell JD, Frey SE, Edwards KM, Keitel WA, Kotloff KL, et al. Effect of varying doses of a monovalent H7N9 influenza vaccine with and without AS03 and MF59 adjuvants on immune response: a randomized clinical trial. JAMA. 2015;314(3):237–46.
Fries LF, Smith GE, Glenn GM. A recombinant viruslike particle influenza A (H7N9) vaccine. N Engl J Med. 2013;369(26):2564–6.
Chung KY, Coyle EM, Jani D, King LR, Bhardwaj R, Fries L, et al. ISCOMATRIX adjuvant promotes epitope spreading and antibody affinity maturation of influenza A H7N9 virus like particle vaccine that correlate with virus neutralization in humans. Vaccine. 2015;33(32):3953–62.
Galson JD, Truck J, Kelly DF, van der Most R. Investigating the effect of AS03 adjuvant on the plasma cell repertoire following pH1N1 influenza vaccination. Sci Rep. 2016;6:37229.
Wong SS, Kaplan B, Zanin M, Debeauchamp J, Kercher L, Crumpton JC, et al. Impact of adjuvants on the immunogenicity and efficacy of split-virion H7N9 vaccine in ferrets. J Infect Dis. 2015;212(4):542–51.
Chen WH, Jackson LA, Edwards KM, Keitel WA, Hill H, Noah DL, et al. Persistence of antibody to influenza A/H5N1 vaccine virus: impact of AS03 adjuvant. Clin Vaccine Immunol. 2015;23(1):73–7.
Oh JZ, Ravindran R, Chassaing B, Carvalho FA, Maddur MS, Bower M, et al. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity. 2014;41(3):478–92.
Taylor DN, Treanor JJ, Sheldon EA, Johnson C, Umlauf S, Song L, et al. Development of VAX128, a recombinant hemagglutinin (HA) influenza-flagellin fusion vaccine with improved safety and immune response. Vaccine. 2012;30(39):5761–9.
Turley CB, Rupp RE, Johnson C, Taylor DN, Wolfson J, Tussey L, et al. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine. 2011;29(32):5145–52.
Jackson DC, Lau YF, Le T, Suhrbier A, Deliyannis G, Cheers C, et al. A totally synthetic vaccine of generic structure that targets Toll-like receptor 2 on dendritic cells and promotes antibody or cytotoxic T cell responses. Proc Natl Acad Sci USA. 2004;101(43):15440–5.
Chua BY, Pejoski D, Turner SJ, Zeng W, Jackson DC. Soluble proteins induce strong CD8+ T cell and antibody responses through electrostatic association with simple cationic or anionic lipopeptides that target TLR2. J Immunol. 2011;187(4):1692–701.
Chua BY, Wong CY, Mifsud EJ, Edenborough KM, Sekiya T, Tan AC, et al. Inactivated influenza vaccine that provides rapid, innate-immune-system-mediated protection and subsequent long-term adaptive immunity. MBio. 2015;6(6):e01024-15.
Sekiya T, Yamagishi J, Gray JHV, Whitney PG, Martinelli A, Zeng W, et al. PEGylation of a TLR2-agonist-based vaccine delivery system improves antigen trafficking and the magnitude of ensuing antibody and CD8+ T cell responses. Biomaterials. 2017;137:61–72.
Nohynek H, Jokinen J, Partinen M, Vaarala O, Kirjavainen T, Sundman J, et al. AS03 adjuvanted AH1N1 vaccine associated with an abrupt increase in the incidence of childhood narcolepsy in Finland. PLoS One. 2012;7(3):e33536.
Miller E, Andrews N, Stellitano L, Stowe J, Winstone AM, Shneerson J, et al. Risk of narcolepsy in children and young people receiving AS03 adjuvanted pandemic A/H1N1 2009 influenza vaccine: retrospective analysis. BMJ. 2013;346:f794.
Wen YM, Mu L, Shi Y. Immunoregulatory functions of immune complexes in vaccine and therapy. EMBO Mol Med. 2016;8(10):1120–33.
Ravetch J. In vivo veritas: the surprising roles of Fc receptors in immunity. Nat Immunol. 2010;11(3):183–5.
Wang TT, Maamary J, Tan GS, Bournazos S, Davis CW, Krammer F, et al. Anti-HA glycoforms drive B cell affinity selection and determine influenza vaccine efficacy. Cell. 2015;162(1):160–9.
Maamary J, Wang TT, Tan GS, Palese P, Ravetch JV. Increasing the breadth and potency of response to the seasonal influenza virus vaccine by immune complex immunization. Proc Natl Acad Sci USA. 2017;114(38):10172–7.
Kilbourne ED, Laver WG, Schulman JL, Webster RG. Antiviral activity of antiserum specific for an influenza virus neuraminidase. J Virol. 1968;2(4):281–8.
Seto JT, Chang FS. Functional significance of sialidase during influenza virus multiplication: an electron microscope study. J Virol. 1969;4(1):58–66.
Schulman JL, Khakpour M, Kilbourne ED. Protective effects of specific immunity to viral neuraminidase on influenza virus infection of mice. J Virol. 1968;2(8):778–86.
McClaren C, Potter CW, Jennings LC. Immunity to influenza in ferrets. XIII. Protection against influenza infection by serum antibody to homologous haemagglutinin or neuraminidase antigens. Med Microbiol Immunol. 1974;160:33–45.
Couch RB, Gordon R, Fedson DS, Kasel JA. Correlated studies of a recombinant influenza-virus vaccine. III. Protection against experimental influenza in man. J Infect Dis. 1971;124:473–80.
Murphy BR, Kasel JA, Chanock RM. Association of serum anti-neuraminidase antibody with resistance to influenza in man. N Engl J Med. 1972;286(25):1329–32.
Monto AS, Kendal AP. Effect of neuraminidase antibody on Hong Kong influenza. Lancet. 1973;1(7804):623–5.
Couch RB, Kasel JA, Gerin JL, Schulman JL, Kilbourne ED. Induction of partial immunity to influenza by a neuraminidase-specific vaccine. J Infect Dis. 1974;129(4):411–20.
Beutner KR, Chow T, Rubi E, Strussenberg J, Clement J, Ogra PL. Evaluation of a neuraminidase specific influenza A virus vaccine in children antibody responses and effects on two successive outbreaks of naturai infection. J Infect Dis. 1979;140:844–50.
Couzens L, Gao J, Westgeest K, Sandbulte M, Lugovtsev V, Fouchier R, et al. An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J Virol Methods. 2014;210C:7–14.
Eichelberger MC, Couzens L, Gao Y, Levine M, Katz J, Wagner R, et al. Comparability of neuraminidase inhibition antibody titers measured by enzyme-linked lectin assay (ELLA) for the analysis of influenza vaccine immunogenicity. Vaccine. 2016;34(4):458–65.
Clements ML, Betts RF, Tierney EL, Murphy BR. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J Clin Microbiol. 1986;24(1):157–60.
Couch RB, Atmar RL, Franco LM, Quarles JM, Wells J, Arden N, et al. Antibody correlates and predictors of immunity to naturally occurring influenza in humans and the importance of antibody to the neuraminidase. J Infect Dis. 2013;207(6):974–81.
Monto AS, Petrie JG, Cross RT, Johnson E, Liu M, Zhong W, et al. Antibody to influenza virus neuraminidase: an independent correlate of protection. J Infect Dis. 2015;212(8):1191–9.
Memoli MJ, Shaw PA, Han A, Czajkowski L, Reed S, Athota R, et al. Evaluation of antihemagglutinin and antineuraminidase antibodies as correlates of protection in an influenza A/H1N1 virus healthy human challenge model. MBio. 2016;7(2):e00417-16.
Sandbulte MR, Westgeest KB, Gao J, Xu X, Klimov AI, Russell CA, et al. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc Natl Acad Sci USA. 2011;108(51):20748–53.
Marcelin G, DuBois R, Rubrum A, Russell CJ, McElhaney JE, Webby RJ. A contributing role for anti-neuraminidase antibodies on immunity to pandemic H1N1 2009 influenza A virus. PLoS One. 2011;6(10):e26335.
Rockman S, Brown LE, Barr IG, Gilbertson B, Lowther S, Kachurin A, et al. Neuraminidase-inhibiting antibody is a correlate of cross-protection against lethal H5N1 influenza virus in ferrets immunized with seasonal influenza vaccine. J Virol. 2013;87(6):3053–61.
Mooney AJ, Gabbard JD, Li Z, Dlugolenski DA, Johnson SK, Tripp RA, et al. Vaccination with recombinant parainfluenza virus 5 expressing neuraminidase protects against homologous and heterologous influenza virus challenge. J Virol. 2017;2017:e00895-17.
Wan H, Gao J, Xu K, Chen H, Couzens LK, Rivers KH, et al. Molecular basis for broad neuraminidase immunity: conserved epitopes in seasonal and pandemic H1N1 as well as H5N1 influenza viruses. J Virol. 2013;87(16):9290–300.
Chen YQ, Wohlbold TJ, Zheng NY, Huang M, Huang Y, Neu KE, et al. Influenza infection in humans induces broadly cross-reactive and protective neuraminidase-reactive antibodies. Cell. 2018;173(2):417–429 e10.
Doyle TM, Hashem AM, Li C, Van Domselaar G, Larocque L, Wang J, et al. Universal anti-neuraminidase antibody inhibiting all influenza A subtypes. Antiviral Res. 2013;100(2):567–74.
Getie-Kebtie M, Sultana I, Eichelberger M, Alterman M. Label-free mass spectrometry-based quantification of hemagglutinin and neuraminidase in influenza virus preparations and vaccines. Influenza Other Respir Viruses. 2013;7(4):521–30.
Kilbourne ED, Cerini CP, Khan MW, Mitchell JWJ, Ogra PL. Immunologic response to the influenza virus neuraminidase is influenced by prior experience with the associated viral heamgglutinin. J Immunol. 1987;138(9):3010–3.
Johansson BE, Bucher DJ, Kilbourne ED. Purified influenza virus hemagglutinin and neuraminidase are equivalent in stimulation of antibody response but induce contrasting types of immunity to infection. J Virol. 1989;63(3):1239–46.
Johansson BE, Kilbourne ED. Dissociation of influenza virus hemagglutinin and neuraminidase eliminates their intravirionic antigenic competition. J Virol. 1993;67(10):5721–3.
Hoskins TW, Davies JR, Smith AJ, Allchin A, Miller CL, Pollock TM. Influenza at Christ’s Hospital: March, 1974. Lancet. 1976;1(7951):105–8.
Kim JH, Liepkalns J, Reber AJ, Lu X, Music N, Jacob J, et al. Prior infection with influenza virus but not vaccination leaves a long-term immunological imprint that intensifies the protective efficacy of antigenically drifted vaccine strains. Vaccine. 2016;34(4):495–502.
Neu KE, Henry Dunand CJ, Wilson PC. Heads, stalks and everything else: how can antibodies eradicate influenza as a human disease? Curr Opin Immunol. 2016;42:48–55.
Wu NC, Wilson IA. A perspective on the structural and functional constraints for immune evasion: insights from influenza virus. J Mol Biol. 2017;429(17):2694–709.
Steel J, Lowen AC, Wang T, Yondola M, Gao Q, Haye K, et al. Influenza virus vaccine based on the conserved hemagglutinin stalk domain. MBio. 2010;1(1):e00018-10.
Krammer F, Pica N, Hai R, Margine I, Palese P. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk-specific antibodies. J Virol. 2013;87(12):6542–50.
Bommakanti G, Lu X, Citron MP, Najar TA, Heidecker GJ, ter Meulen J, et al. Design of Escherichia coli-expressed stalk domain immunogens of H1N1 hemagglutinin that protect mice from lethal challenge. J Virol. 2012;86(24):13434–44.
Impagliazzo A, Milder F, Kuipers H, Wagner MV, Zhu X, Hoffman RM, et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science. 2015;349(6254):1301–6.
Mallajosyula VV, Citron M, Ferrara F, Lu X, Callahan C, Heidecker GJ, et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc Natl Acad Sci U S A. 2014;111(25):E2514–23.
Mallajosyula VV, Citron M, Ferrara F, Temperton NJ, Liang X, Flynn JA, et al. Hemagglutinin sequence conservation guided stem immunogen design from influenza A H3 subtype. Front Immunol. 2015;6:329.
Yassine HM, Boyington JC, McTamney PM, Wei CJ, Kanekiyo M, Kong WP, et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med. 2015;21(9):1065–70.
Sutton TC, Chakraborty S, Mallajosyula VVA, Lamirande EW, Ganti K, Bock KW, et al. Protective efficacy of influenza group 2 hemagglutinin stem-fragment immunogen vaccines. NPJ Vaccines. 2017;2:35.
Yap KL, Ada GL. The recovery of mice from influenza A virus infection: adoptive transfer of immunity with influenza virus-specific cytotoxic T lymphocytes recognizing a common virion antigen. Scand J Immunol. 1978;8(5):413–20.
Flynn KJ, Belz GT, Altman JD, Ahmed R, Woodland DL, Doherty PC. Virus-specific CD8+ T cells in primary and secondary influenza pneumonia. Immunity. 1998;8(6):683–91.
Slutter B, Pewe LL, Lauer P, Harty JT. Cutting edge: rapid boosting of cross-reactive memory CD8 T cells broadens the protective capacity of the Flumist vaccine. J Immunol. 2013;190(8):3854–8.
Baz M, Boonnak K, Paskel M, Santos C, Powell T, Townsend A, et al. Nonreplicating influenza A virus vaccines confer broad protection against lethal challenge. MBio. 2015;6(5):e01487-15.
Li J, Arevalo MT, Chen Y, Chen S, Zeng M. T-cell-mediated cross-strain protective immunity elicited by prime-boost vaccination with a live attenuated influenza vaccine. Int J Infect Dis. 2014;27:37–43.
He XS, Holmes TH, Zhang C, Mahmood K, Kemble GW, Lewis DB, et al. Cellular immune responses in children and adults receiving inactivated or live attenuated influenza vaccines. J Virol. 2006;80(23):11756–66.
Basha S, Hazenfeld S, Brady RC, Subbramanian RA. Comparison of antibody and T-cell responses elicited by licensed inactivated- and live-attenuated influenza vaccines against H3N2 hemagglutinin. Hum Immunol. 2011;72(6):463–9.
Hoft DF, Babusis E, Worku S, Spencer CT, Lottenbach K, Truscott SM, et al. Live and inactivated influenza vaccines induce similar humoral responses, but only live vaccines induce diverse T-cell responses in young children. J Infect Dis. 2011;204(6):845–53.
Mohn KG, Bredholt G, Brokstad KA, Pathirana RD, Aarstad HJ, Tondel C, et al. Longevity of B-cell and T-cell responses after live attenuated influenza vaccination in children. J Infect Dis. 2015;211(10):1541–9.
Belshe R, Lee MS, Walker RE, Stoddard J, Mendelman PM. Safety, immunogenicity and efficacy of intranasal, live attenuated influenza vaccine. Expert Rev Vaccines. 2004;3(6):643–54.
Clover RD, Crawford S, Glezen WP, Taber LH, Matson CC, Couch RB. Comparison of heterotypic protection against influenza A/Taiwan/86 (H1N1) by attenuated and inactivated vaccines to A/Chile/83-like viruses. J Infect Dis. 1991;163(2):300–4.
Tricco AC, Chit A, Soobiah C, Hallett D, Meier G, Chen MH, et al. Comparing influenza vaccine efficacy against mismatched and matched strains: a systematic review and meta-analysis. BMC Med. 2013;11:153.
Steinhoff MC, Fries LF, Karron RA, Clements ML, Murphy BR. Effect of heterosubtypic immunity on infection with attenuated influenza A virus vaccines in young children. J Clin Microbiol. 1993;31(4):836–8.
Grohskopf LA, Sokolow LZ, Fry AM, Walter EB, Jernigan DB. Update: ACIP recommendations for the use of quadrivalent live attenuated influenza vaccine (LAIV4)—United States, 2018–19 influenza season. MMWR Morb Mortal Wkly Rep. 2018;67(22):643–5.
Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibility complex-restricted CD8+ T cells. J Exp Med. 1991;174(4):875–80.
Epstein SL, Lo CY, Misplon JA, Lawson CM, Hendrickson BA, Max EE, et al. Mechanisms of heterosubtypic immunity to lethal influenza A virus infection in fully immunocompetent, T cell-depleted, beta2-microglobulin-deficient, and J chain-deficient mice. J Immunol. 1997;158(3):1222–30.
Topham DJ, Doherty PC. Clearance of an influenza A virus by CD4+ T cells is inefficient in the absence of B cells. J Virol. 1998;72(1):882–5.
Yap KL, Ada GL, McKenzie IF. Transfer of specific cytotoxic T lymphocytes protects mice inoculated with influenza virus. Nature. 1978;273(5659):238–9.
Bender BS, Croghan T, Zhang L, Small PA Jr. Transgenic mice lacking class I major histocompatibility complex-restricted T cells have delayed viral clearance and increased mortality after influenza virus challenge. J Exp Med. 1992;175(4):1143–5.
McKinstry KK, Strutt TM, Kuang Y, Brown DM, Sell S, Dutton RW, et al. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Investig. 2012;122(8):2847–56.
Wilkinson TM, Li CK, Chui CS, Huang AK, Perkins M, Liebner JC, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med. 2012;18(2):274–80.
Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, et al. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med. 2013;19(10):1305–12.
Hayward AC, Wang L, Goonetilleke N, Fragaszy EB, Bermingham A, Copas A, et al. Natural T cell-mediated protection against seasonal and pandemic influenza. Results of the Flu Watch Cohort Study. Am J Respir Crit Care Med. 2015;191(12):1422–31.
Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity. 2011;34(1):108–21.
Alam S, Knowlden ZA, Sangster MY, Sant AJ. CD4 T cell help is limiting and selective during the primary B cell response to influenza virus infection. J Virol. 2014;88(1):314–24.
Leddon SA, Richards KA, Treanor JJ, Sant AJ. Abundance and specificity of influenza reactive circulating memory follicular helper and non-follicular helper CD4 T cells in healthy adults. Immunology. 2015;146(1):157–62.
Bentebibel SE, Lopez S, Obermoser G, Schmitt N, Mueller C, Harrod C, et al. Induction of ICOS+CXCR3+CXCR5+ TH cells correlates with antibody responses to influenza vaccination. Sci Transl Med. 2013;5(176):176ra32.
Herati RS, Muselman A, Vella L, Bengsch B, Parkhouse K, Del Alcazar D, et al. Successive annual influenza vaccination induces a recurrent oligoclonotypic memory response in circulating T follicular helper cells. Sci Immunol. 2017;2(8):eaag2152.
Chen L, Zanker D, Xiao K, Wu C, Zou Q, Chen W. Immunodominant CD4+ T-cell responses to influenza A virus in healthy individuals focus on matrix 1 and nucleoprotein. J Virol. 2014;88(20):11760–73.
Pilkinton MA, Nicholas KJ, Warren CM, Smith RM, Yoder SM, Talbot HK, et al. Greater activation of peripheral T follicular helper cells following high dose influenza vaccine in older adults forecasts seroconversion. Vaccine. 2017;35(2):329–36.
Wiley JA, Cerwenka A, Harkema JR, Dutton RW, Harmsen AG. Production of interferon-gamma by influenza hemagglutinin-specific CD8 effector T cells influences the development of pulmonary immunopathology. Am J Pathol. 2001;158(1):119–30.
Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 2001;291(5512):2413–7.
Klonowski KD, Williams KJ, Marzo AL, Blair DA, Lingenheld EG, Lefrancois L. Dynamics of blood-borne CD8 memory T cell migration in vivo. Immunity. 2004;20(5):551–62.
Clark RA, Chong B, Mirchandani N, Brinster NK, Yamanaka K, Dowgiert RK, et al. The vast majority of CLA+ T cells are resident in normal skin. J Immunol. 2006;176(7):4431–9.
Wakim LM, Gebhardt T, Heath WR, Carbone FR. Cutting edge: local recall responses by memory T cells newly recruited to peripheral nonlymphoid tissues. J Immunol. 2008;181(9):5837–41.
Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol. 2009;10(5):524–30.
Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. 2014;95(2):215–24.
Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol. 2012;188(10):4866–75.
Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One. 2011;6(1):e16245.
Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38(1):187–97.
Wakim LM, Smith J, Caminschi I, Lahoud MH, Villadangos JA. Antibody-targeted vaccination to lung dendritic cells generates tissue-resident memory CD8 T cells that are highly protective against influenza virus infection. Mucosal Immunol. 2015;8(5):1060–71.
Zens KD, Chen JK, Farber DL. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight. 2016;1(10):e85832.
McMichael AJ, Gotch FM, Dongworth DW, Clark A, Potter CW. Declining T-cell immunity to influenza, 1977–82. Lancet. 1983;2(8353):762–4.
van de Sandt CE, Hillaire ML, Geelhoed-Mieras MM, Osterhaus AD, Fouchier RA, Rimmelzwaan GF. Human influenza A virus-specific CD8+ T-cell response is long-lived. J Infect Dis. 2015;212(1):81–5.
Nogales A, Baker SF, Domm W, Martinez-Sobrido L. Development and applications of single-cycle infectious influenza A virus (sciIAV). Virus Res. 2016;216:26–40.
Powell TJ, Silk JD, Sharps J, Fodor E, Townsend AR. Pseudotyped influenza A virus as a vaccine for the induction of heterotypic immunity. J Virol. 2012;86(24):13397–406.
Katsura H, Iwatsuki-Horimoto K, Fukuyama S, Watanabe S, Sakabe S, Hatta Y, et al. A replication-incompetent virus possessing an uncleavable hemagglutinin as an influenza vaccine. Vaccine. 2012;30(42):6027–33.
Guo H, Baker SF, Martinez-Sobrido L, Topham DJ. Induction of CD8 T cell heterologous protection by a single dose of single-cycle infectious influenza virus. J Virol. 2014;88(20):12006–16.
Sarawar S, Hatta Y, Watanabe S, Dias P, Neumann G, Kawaoka Y, et al. M2SR, a novel live single replication influenza virus vaccine, provides effective heterosubtypic protection in mice. Vaccine. 2016;34(42):5090–8.
Hatta Y, Boltz D, Sarawar S, Kawaoka Y, Neumann G, Bilsel P. M2SR, a novel live influenza vaccine, protects mice and ferrets against highly pathogenic avian influenza. Vaccine. 2017;35(33):4177–83.
Si L, Xu H, Zhou X, Zhang Z, Tian Z, Wang Y, et al. Generation of influenza A viruses as live but replication-incompetent virus vaccines. Science. 2016;354(6316):1170–3.
Mueller S, Coleman JR, Papamichail D, Ward CB, Nimnual A, Futcher B, et al. Live attenuated influenza virus vaccines by computer-aided rational design. Nat Biotechnol. 2010;28(7):723–6.
Broadbent AJ, Santos CP, Anafu A, Wimmer E, Mueller S, Subbarao K. Evaluation of the attenuation, immunogenicity, and efficacy of a live virus vaccine generated by codon-pair bias de-optimization of the 2009 pandemic H1N1 influenza virus, in ferrets. Vaccine. 2016;34(4):563–70.
Cox A, Baker SF, Nogales A, Martinez-Sobrido L, Dewhurst S. Development of a mouse-adapted live attenuated influenza virus that permits in vivo analysis of enhancements to the safety of live attenuated influenza virus vaccine. J Virol. 2015;89(6):3421–6.
Morgan SB, Hemmink JD, Porter E, Harley R, Shelton H, Aramouni M, et al. Aerosol delivery of a candidate universal influenza vaccine reduces viral load in pigs challenged with pandemic H1N1 virus. J Immunol. 2016;196(12):5014–23.
Holzer B, Morgan SB, Matsuoka Y, Edmans M, Salguero FJ, Everett H, et al. Comparison of heterosubtypic protection in ferrets and pigs induced by a single-cycle influenza vaccine. J Immunol. 2018;200(12):4068–77.
Gotch F, McMichael A, Smith G, Moss B. Identification of viral molecules recognized by influenza-specific human cytotoxic T lymphocytes. J Exp Med. 1987;165(2):408–16.
Gianfrani C, Oseroff C, Sidney J, Chesnut RW, Sette A. Human memory CTL response specific for influenza A virus is broad and multispecific. Hum Immunol. 2000;61(5):438–52.
Boon AC, de Mutsert G, van Baarle D, Smith DJ, Lapedes AS, Fouchier RA, et al. Recognition of homo- and heterosubtypic variants of influenza A viruses by human CD8+ T lymphocytes. J Immunol. 2004;172(4):2453–60.
Lee LY, Ha do LA, Simmons C, de Jong MD, Chau NV, Schumacher R, et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J Clin Investig. 2008;118(10):3478–90.
Stewart-Jones GB, McMichael AJ, Bell JI, Stuart DI, Jones EY. A structural basis for immunodominant human T cell receptor recognition. Nat Immunol. 2003;4(7):657–63.
Bui HH, Peters B, Assarsson E, Mbawuike I, Sette A. Ab and T cell epitopes of influenza A virus, knowledge and opportunities. Proc Natl Acad Sci USA. 2007;104(1):246–51.
Grant E, Wu C, Chan KF, Eckle S, Bharadwaj M, Zou QM, et al. Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T-cell responses. Immunol Cell Biol. 2013;91(2):184–94.
Quinones-Parra S, Grant E, Loh L, Nguyen TH, Campbell KA, Tong SY, et al. Preexisting CD8+ T-cell immunity to the H7N9 influenza A virus varies across ethnicities. Proc Natl Acad Sci USA. 2014;111(3):1049–54.
Grant EJ, Chen L, Quinones-Parra S, Pang K, Kedzierska K, Chen W. T-cell immunity to influenza A viruses. Crit Rev Immunol. 2014;34(1):15–39.
Pleguezuelos O, Robinson S, Stoloff GA, Caparros-Wanderley W. Synthetic influenza vaccine (FLU-v) stimulates cell mediated immunity in a double-blind, randomised, placebo-controlled Phase I trial. Vaccine. 2012;30(31):4655–60.
Francis JN, Bunce CJ, Horlock C, Watson JM, Warrington SJ, Georges B, et al. A novel peptide-based pan-influenza A vaccine: a double blind, randomised clinical trial of immunogenicity and safety. Vaccine. 2015;33(2):396–402.
Atsmon J, Caraco Y, Ziv-Sefer S, Shaikevich D, Abramov E, Volokhov I, et al. Priming by a novel universal influenza vaccine (Multimeric-001)—a gateway for improving immune response in the elderly population. Vaccine. 2014;32(44):5816–23.
Lowell GH, Ziv S, Bruzil S, Babecoff R, Ben-Yedidia T. Back to the future: Immunization with M-001 prior to trivalent influenza vaccine in 2011/12 enhanced protective immune responses against 2014/15 epidemic strain. Vaccine. 2017;35(5):713–5.
Rosendahl Huber S, van Beek J, de Jonge J, Luytjes W, van Baarle D. T cell responses to viral infections - opportunities for Peptide vaccination. Front Immunol. 2014;5:171.
Ura T, Okuda K, Shimada M. Developments in viral vector-based vaccines. Vaccines (Basel). 2014;2(3):624–41.
Lillie PJ, Berthoud TK, Powell TJ, Lambe T, Mullarkey C, Spencer AJ, et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin Infect Dis. 2012;55(1):19–25.
Antrobus RD, Berthoud TK, Mullarkey CE, Hoschler K, Coughlan L, Zambon M, et al. Coadministration of seasonal influenza vaccine and MVA-NP+M1 simultaneously achieves potent humoral and cell-mediated responses. Mol Ther. 2014;22(1):233–8.
Tully CM, Chinnakannan S, Mullarkey CE, Ulaszewska M, Ferrara F, Temperton N, et al. Novel bivalent viral-vectored vaccines induce potent humoral and cellular immune responses conferring protection against stringent influenza A virus challenge. J Immunol. 2017;199(4):1333–41.
Florek NW, Weinfurter JT, Jegaskanda S, Brewoo JN, Powell TD, Young GR, et al. Modified vaccinia virus Ankara encoding influenza virus hemagglutinin induces heterosubtypic immunity in macaques. J Virol. 2014;88(22):13418–28.
Talaat KR, Luke CJ, Khurana S, Manischewitz J, King LR, McMahon BA, et al. A live attenuated influenza A(H5N1) vaccine induces long-term immunity in the absence of a primary antibody response. J Infect Dis. 2014;209(12):1860–9.
Pitisuttithum P, Boonnak K, Chamnanchanunt S, Puthavathana P, Luvira V, Lerdsamran H, et al. Safety and immunogenicity of a live attenuated influenza H5 candidate vaccine strain A/17/turkey/Turkey/05/133 H5N2 and its priming effects for potential pre-pandemic use: a randomised, double-blind, placebo-controlled trial. Lancet Infect Dis. 2017;17(8):833–42.
Ledgerwood JE, Wei C-J, Hu Z, Gordon IJ, Enama ME, Hendel CS, et al. DNA priming and influenza vaccine immunogenicity: two phase 1 open label randomised clinical trials. Lancet Infect Dis. 2011;11(12):916–24.
Sobhanie M, Matsuoka Y, Jegaskanda S, Fitzgerald T, Mallory R, Chen Z, et al. Evaluation of the safety and immunogenicity of a candidate pandemic live attenuated influenza vaccine (pLAIV) against influenza A(H7N9). J Infect Dis. 2016;213(6):922–9.
Jegaskanda S, Mason RD, Andrews SF, Wheatley AK, Zhang R, Reynoso GV, et al. Intranasal live influenza vaccine priming elicits localized B cell responses in mediastinal lymph nodes. J Virol. 2018;92(9):e01970-17.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this manuscript.
Conflict of interest
A. Fox, K. Quinn and K. Subbarao declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Fox, A., Quinn, K.M. & Subbarao, K. Extending the Breadth of Influenza Vaccines: Status and Prospects for a Universal Vaccine. Drugs 78, 1297–1308 (2018). https://doi.org/10.1007/s40265-018-0958-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0958-7