
Abstract
Craniosynostosis, a condition that includes the premature fusion of one or multiple cranial sutures, is a relatively common birth defect in humans and the second most common craniofacial anomaly after orofacial clefts. There is a significant clinical variation among different sutural synostoses as well as significant variation within any given single-suture synostosis. Craniosynostosis can be isolated (i.e., nonsyndromic) or occurs as part of a genetic syndrome (e.g., Crouzon, Pfeiffer, Apert, Muenke, and Saethre–Chotzen syndromes). Approximately 85 % of all cases of craniosynostosis are nonsyndromic. Several recent genomic discoveries are elucidating the genetic basis for nonsyndromic cases and implicate the newly identified genes in signaling pathways previously found in syndromic craniosynostosis. Published epidemiologic and phenotypic studies clearly demonstrate that nonsyndromic craniosynostosis is a complex and heterogeneous condition supporting a strong genetic component accompanied by environmental factors that contribute to the pathogenetic network of this birth defect. Large population, rather than single-clinic or hospital-based studies is required with phenotypically homogeneous subsets of patients to further understand the complex genetic, maternal, environmental, and stochastic factors contributing to nonsyndromic craniosynostosis. Learning about these variables is a key in formulating the basis of multidisciplinary and lifelong care for patients with these conditions.
Similar content being viewed by others


Introduction
Craniosynostosis, which always involves the premature fusion of one or more of the neurocranial sutures and can include many associated dysmorphologies of the craniofacial complex, is a relatively common congenital malformation [1]. The incidence of craniosynostosis is estimated to be in the range of 1 in 2,000–2,500 live births and occurs in all ethnic groups [2–5]. Approximately 85 % of cases are nonsyndromic, and 92 % are non-familial. The other 15 % of cases are defined as having one of the more than 180 known craniosynostosis syndromes, at least 50 % of which follow a Mendelian pattern of inheritance [6, 7].
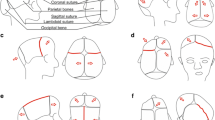
The frequency of fusion of each of the cranial vault sutures varies. Sagittal synostosis, the most common of the isolated craniosynostoses, occurs in 45–58 % of all craniosynostoses with males more often affected than females (M:F ratio of 3.5:1) [3, 8]. The fusion of the midline sagittal suture results in scaphocephaly, a skull shape that is relatively longer than normal along the anterior-posterior axis and narrowed mediolaterally (Fig. 1a). However, there is a great variability in the scaphocephalic morphology of sagittal synostosis that involves not only the cranial vault but also the facial skeleton and cranial base [9]. Little is known about the sources of this heterogeneity.

Variation in cranial vault shape depicted by three-dimensional computed tomography (CT) reconstructions of infants with various types of single-suture craniosynostosis (superior and inferior (i.e., ectocranial) views; anterior aspect of skull at top, posterior aspect of skull at bottom). Those pictured are cases of single-suture craniosynostosis from our archive of 3D CT images and include skulls of infants with a sagittal craniosynostosis; b bicoronal, right unicoronal, and left unicoronal craniosynostoses (from top to bottom); c metopic craniosynostosis; d bilateral, right unilateral, and left unilateral lambdoidal craniosynostoses (from top to bottom); e unaffected individual. The black arrow heads indicate the sutures that are prematurely closed. Though most attention has been focused on cranial vault shape, the cranial base and facial skeleton are also dysmorphic in craniosynostosis conditions. Not to scale
Coronal synostosis occurs in 20–30 % of all cases of craniosynostosis with females more often affected than males (M:F 1:2) [10, 11]. The overt cranial dysmorphology of coronal craniosynostosis varies depending upon whether premature closure of the coronal suture occurs bilaterally involving both the right and the left side of the skull (resulting in brachycephaly) or unilaterally (resulting in anterior plagiocephaly or asymmetry) (Fig. 1b). Unilateral coronal synostosis occurs twice as often as bilateral coronal synostosis. Progressive frontal plagiocephaly or flattening sometimes results from the fusion of the fronto-sphenoidal or fronto-zygomatic sutures and is detected by the detailed three-dimensional computed tomography (3D-CT) imaging of the basilar coronal ring sutures involving the ethmoid-sphenoidal sutures [12, 13]. Metopic synostosis, resulting in trigonocephaly (Fig. 1c), had an estimated prevalence of 6–7 in 100,000 live births prior to 2000, but in the past decade its presentation in some medical centers in Europe and the US has increased as much as fourfold for unknown reasons [14, 15]. Approximately 67 % of all metopic cases are nonsyndromic, and 92 % are non-familial [16]. Male to female ratio has been estimated to be about 3:1. Lambdoid synostosis, resulting in posterior plagiocephaly (Fig. 1d), is estimated to represent about 1 % of all craniosynostosis [1].
Candidate Gene Mutations Found in Nonsyndromic Craniosynostosis
Causative mutations for craniosynostosis have been primarily identified in coronal craniosynostosis syndromes most often within the FGFR1, FGFR2, FGFR3, TWIST1, and EFNB1 genes, but the list of new genes involved in less common syndromes is growing (Table 1), reviewed by Passos-Bueno et al. [17], Wilkie et al. [18], and Jabs and Lewanda [19], and there is an association between the pattern of facial dysmorphogenesis and causative mutation for some of these syndromes [20∙]. However, the genetic etiology of nonsyndromic craniosynostosis (NSC) remained poorly understood until very recently. Over the last two decades, the search for genetic mutations underlying NSC has focused on "hotspots" of genes that are known to cause syndromic craniosynostosis [21]. Rare mutations in FGFRs, TWIST1, LRIT3, ALX4, IGFR1, EFNA4, RUNX2, and FREM1 have been reported in a minor fraction of NSC cases (Table 1).
An example of a successful identification of a single point mutation in a candidate gene is the fibroblast growth factor receptor 3 (FGFR3) Pro250Arg mutation associated with individuals initially diagnosed with isolated coronal craniosynostosis [22, 23]. The identification of the FGFR3 Pro250Arg mutation resulted in the definition of Muenke syndrome [23] characterized by a highly variable phenotype with some individuals appearing phenotypically normal [24] demonstrating a reduced penetrance of the mutation at about 80 % [18]. It has been estimated that the FGFR3 Pro250Arg mutation may account for 4–12 % of isolated unilateral and 30–40 % of isolated bilateral coronal synostosis cases [25, 26] with a population prevalence of about 1 case per 30,000 [18].
Other rare gene mutations have been identified for isolated synostosis [27]. However, some of these mutations were present not only in the affected probands, but also in other members of the family, who had craniofacial dysmorphisms (but not craniosynostosis) or were unaffected suggesting incomplete penetrance [28–32]. In one case of sagittal NSC, the FGFR2 Ala315Thr mutation was reported [33]. Two cases with sagittal NSC were found to carry Ser494Thr and Cys592Tyr mutations in LRIT3, a protein believed to regulate maturation and signaling of FGFR1 [28]; another three sagittal NSC cases had Val7Phe, Lys211Glu, and Pro306Leu mutations in ALX4, a homeobox containing transcription factor regulating calvarial development through interactions with Wnt and bone morphogenetic proteins (BMPs) [31]. TWIST1 mutations, Ala186Thr, Ser201Tyr, and Ser188Leu in the TWIST Box domain, were found in two cases of the isolated sagittal synostosis and in one case of isolated left coronal synostosis, respectively [29, 30]. Insulin-like growth factor 1 receptor (IGF1R) mutations, R406H, N857S, and R595H, were found in two cases of isolated sagittal and one with coronal synostosis, and rare variants P190S and M446V were also detected [34]. For coronal NSC, an FGFR2 Ala315Ser mutation was reported in a patient with unicoronal synostosis and a birth history of breech presentation and skull compression [35]. EFNA4 His60Tyr, Pro117Thr, and Asn157LysfsX45 mutations have been reported in three patients with coronal NSC [32]. An FGFR1 Ile1300Trp mutation was found in one case of metopic NSC with facial skin tags [36]. Recently, a 1.1 Mb duplication encompassing RUNX2 and mutations in FREM1 has been associated with metopic NSC [37, 38]. Apart from IGFR1 and FREM1, the above genes can be linked directly to TWIST1 and the FGF signaling pathway, which ultimately interact to control the entry of mesenchymal cells into osteoblastic differentiation in the developing suture; it is speculated that FREM1 may also bind FGFs to modulate the FGF pathway [38].
Novel Genes Identified in Craniosynostosis Using Genomic Technologies
In the past 2 years, important breakthroughs have been achieved with the identification of new genes associated with sagittal and coronal NSC. Justice et al. [39∙∙] reported susceptibility loci for sagittal NSC near BMP2 and BBS9. Twigg et al. [40∙∙] showed that the reduced dosage of ERF causes sagittal, lambdoid, and multisuture craniosynostoses in cases diagnosed as isolated or syndromic craniosynostosis. Finally, Sharma et al. [41∙∙] identified mutations within TCF12 associated with unilateral and bilateral coronal craniosynostosis in patients with isolated or syndromic craniosynostosis. Here, we review these mutations in genes that may account for a significant number of patients diagnosed with NSC.
Susceptibility Loci for Sagittal NSC Near BMP2 and BBS9
Though sagittal NSC is the most frequent form of craniosynostosis, the genetic basis for most cases is unknown, and only rare gene mutations have been identified until recently [27]. Justice et al. [39∙∙] conducted a genome wide association study (GWAS) of 130 non-Hispanic case-parent trios of European ancestry followed by the replication analysis of 172 unrelated non-Hispanic Caucasian cases and 548 controls to identify susceptibility loci for sagittal NSC near BMP2 and within BBS9. The discovery/replication meta-analysis demonstrated the combined odds ratios of 4.38 (95 % CI 3.51–5.45; P = 1.1 × 10−39) and 0.24 (95 % CI 0.17–0.32; P = 5.6 × 10−20), respectively. BMP2 is a member of the TGF-β superfamily and a key growth factor regulating osteoblast development [42]. The BMP and FGF pathways interact and are important in skull growth [43–45]. BBS9 is a member of the BBSome, a multiprotein complex localized in the primary cilium that is involved in coordinating many developmentally important signaling pathways including platelet-derived growth factor receptor α, sonic hedgehog, and Wnt [46, 47]. The BBSome is also implicated in intraflagellar transport [48]. BBS9 loss-of-function mutations have been found in Bardet–Biedl syndrome (BBS) patients [49]. Although BBS-affected individuals do not present with suture phenotypes, there are ciliopathy conditions that have craniosynostosis as a feature such as cranioectodermal dysplasia (Sensenbrenner syndrome) [50–53].
As of this writing, no study phenotypically characterizing sagittal NSC cases with or without BMP2 or BBS9 variations has been published. Although the morphology of the cranial vault has been observed as a defining characteristic in craniosynostosis, qualitative assessments of calvarial dysmorphology have shown consistent variability in NSC (Fig. 1) [54], and the exact source of this variation remains unknown. A recent quantitative study of craniofacial shape in 43 infants with nonsyndromic sagittal synostosis using 3D-CT reconstruction and morphometric methods confirmed variation in cranial vault morphology [9]. In all cases studied, the central portion of the sagittal suture was the first to fuse (probably prenatally), and at least two different developmental paths toward complete fusion of the sagittal suture exist either in the anterior section or in the posterior section of the sagittal suture being the second to fuse. The analyses showed association between the variation in craniofacial shape and the exact path of fusion of the sagittal suture. Comparable morphometric studies should be completed using cases carrying either BMP2 or BBS9 NSC-associated variants to determine whether or not these genetic influences correspond with specific phenotypes.
ERF and TCF12 Mutations in Patients with Craniosynostosis
Using whole exome sequencing of seven unrelated individuals with bilateral coronal synostosis and negative for previously described mutations [55], two additional genes, ERF and TCF12, were found to have mutations in two or more patients with craniosynostosis [40∙∙, 41∙∙]. Heterozygous mutations in ERF, an inhibitory ETS family transcription factor that is negatively regulated by the extracellular signal-related kinases 1 and 2 (ERK1/2) of the mitogen-activated protein kinase (MAPK) signaling pathway, were found [40∙∙]. Some of the patients with the ERF mutations were syndromic with sagittal or multisuture synostosis, craniofacial dysmorphology, Chiari malformation, and language delay, and others were nonsyndromic with sagittal, unilateral or bilateral lambdoid, and multisuture synostosis. ERF was found to bind close to regulatory sites recognized by RUNX2, an essential regulator of osteoblast differentiation, and could interfere with transcriptional activation by RUNX2. Thus, loss-of-function mutations of ERF in these patients have a similar effect to FGFR-phosphorylated ERK activation observed in FGFR-related craniosynostoses. Reduced ERF function in these conditions can result in the upregulation of RUNX2 activity, leading to changes in osteoblast differentiation and potential premature ossification of cranial sutures.
Heterozygous mutations in TCF12, transcription factor 12, were also identified in syndromic and nonsyndromic patients with unilateral and bilateral coronal and multisuture synostoses [41∙∙, 56]. Syndromic patients had additional features of craniofacial dysmorphism and external ear and minor limb anomalies. TCF12 mutations were found in 32 % of subjects with bilateral and 10 % with unilateral coronal synostosis. TCF12 heterodimerizes with class II basic helix-loop-helix transcription factors including TWIST1. Loss-of-function mutations in TWIST1 have previously been shown to cause the Saethre–Chotzen syndrome, a craniosynostosis condition with coronal fusion and minor limb anomalies [57, 58]. The TCF12-TWIST1 heterodimer is likely to regulate the specification of the coronal suture between the neural crest-derived frontal bones and mesoderm-derived parietal bones [32, 59]. As in the case of ERF, these dimers may inhibit osteogenic differentiation via actions on RUNX2 and FGFR signaling pathways [60].
Genetic Risk
To estimate the proportion of craniosynostosis patients that screen positive for a gene mutation, a study was conducted on 326 children, who were born from 1993 to 2002 and required surgical treatment in a craniofacial unit in Oxford, England [18]. Genetic diagnoses were made for 21 % of all craniosynostosis cases, and the FGFR3 P250R mutation was the single most common mutation, accounting for 24 % of cases with genetic diagnoses (5 % of all cases). Those with genetic diagnoses were associated with increased rates of many complications. Children with the clinical diagnosis of nonsyndromic unicoronal or bicoronal synostosis were more likely to have an identified causative mutation than those with other sutural involvement. In the extended Oxford birth cohort (cases born from 1998 to 2006), TCF12 mutations were identified in approximately 1.0 % of craniosynostosis cases [41]. While the patients with TCF12 mutations had a more benign course than patients with FGFR3 P250R or TWIST1 mutations, 14 % had developmental delay or learning disabilities and two were diagnosed with autism. ERF mutations explain an additional 1.2 % of etiology for the cohort [40].
Cellular and Animal Model Investigations of Craniosynostosis
Both the genome wide expression analysis of primary osteoblasts derived from craniosynostosis patients and the creation of mutant mouse models allow experimental analysis of craniosynostosis phenotypes and the roles of newly-discovered genes in craniosynostosis. Gene expression profiling of human craniosynostosis samples has been recently reviewed [61]. As an example, a recent survey of 199 NSC patient-derived osteoblasts, including sagittal, metopic, and coronal cases, suggested the common involvement of FGF7, SFRP4, and VCAM1 and the role of extracellular matrix interactions in the craniosynostosis phenotypes [62].
Mouse models of activating Fgfr mutations and of Twist1 loss-of-function have been invaluable in understanding the coronal synostosis and the role of the neural crest/mesoderm boundary forming this suture [63∙, 64]. Erf was shown to be expressed within calvarial sutures in the mouse, and the conditional deletion of Erf demonstrated that loss of Erf was causative for craniosynostosis [40]. While heterozygous null Tcf12 mice alone did not show craniosynostosis, reduction of Tcf12 significantly increased the incidence and severity of craniosynostosis in Twist1 heterozygous null mice, supporting the model of Tcf12 interaction with Twist1 [41]. Gene expression within sutures can be readily determined in mice, and this knowledge is crucial in understanding the connection between gene mutations and specific patterns of suture fusion or other resulting craniofacial dysmorphologies, exemplified by Fgfrs1–3 and Twist1, which have distinct expression patterns within sutures [65]. The restriction of craniosynostosis to specific sutures may reflect the suture-specific expression of some genes. For example, in the mouse model of Greig cephalopolysyndactyly syndrome, the Gli3 (Xt-J/Xt-J) mouse, the lambdoid sutures fuse and interfrontal suture development is anomalous, and these are the sites of strong embryonic calvarial Gli3 expression [66]. A comprehensive knowledge of murine sutural gene expression, particularly at embryonic stages, would facilitate the identification of human craniosynostosis candidate genes. Furthermore, the mouse provides a model for the potential therapeutic amelioration of craniosynostosis. Chemical inhibition of Fgfr tyrosine kinase activity, or of the activity of effector kinases downstream of Fgfrs, results in reduced craniosynostosis in mouse models of Crouzon, Apert, and Beare-Stevenson syndromes [67–70].
Conclusions
The etiology of approximately three quarters of patients diagnosed with craniosynostosis is not known. Thus, much work is needed in the elucidation of causal mutations proximate to the BMP2 and BBS9 gene loci and in identifying new genes involved in craniosynostosis. Since craniosynostosis is a complex heterogeneous condition, the interplay between genetic variants and environmental exposures may explain the low heritability for NSC, and their combined action may elucidate processes underlying the variable dysmorphogenesis of the facial skeleton and cranial base, portions of the skull that do not encompass the cranial vault sutures. These factors may also contribute to the lack of distinct disease phenotypes across diagnostic groups and the current lack of identified molecular causes in some cases. Mutations in genes initially identified in syndromic cases may contribute to causation in milder phenotypes including nonsyndromic or non-penetrant cases. The large population of patients with nonsyndromic craniosynostosis embodies a fundamental need for more work, as well as a fertile research area for the discovery of novel genetic, maternal, and environmental factors. Harnessing next generation sequencing technology and bioinformatics analysis with our understanding of the genome, transcriptome, and epigenome will help to elucidate the etiology of craniosynostosis, both syndromic and nonsyndromic.
As noted previously, although sutural fusion is the most frequent feature studied and treated, craniosynostosis also refers to the abnormal development of the bones of the skull associated with dysmorphic skull shape. In animal models for human craniosynostosis syndromes, the abnormal skull shape can be detected before the premature closure of cranial vault sutures 71∙, 72∙]. The development of animal models for craniosynostosis [70, 73, 74] has already revealed many molecularly driven three-dimensional morphological changes in soft tissues of the head and skull that were not apparent in humans [71∙, 75, 76∙, 77]. These changes are more difficult to evaluate quantitatively in humans where observations are routinely made postnatally and there is a lack of appropriate morphological control data sets to make meaningful comparisons to abnormal phenotypes. Human cases provide access to population-based molecular screens, and more recently the chance to link genotype with phenotype [20], but do not provide easy access to the molecularly-based processes that result in the highly variable, abnormal cranial morphology of syndromic and nonsyndromic cases of craniosynostosis. This access will be necessary in order to understand what unites the various molecular causes of craniosynostosis (Table 1) at the genomic and phenotypic levels. Mouse models provide access to both the processes that underlie these integrated sets of anomalies and the networks that produce them. Emerging technologies (e.g., optical projection tomography [78]) allow direct study of the correspondence between the spatiotemporal dynamics of gene expression patterns, morphogenesis, and morphological diversity.
The coordinated assimilation of results from human- and animal model-based research that build on respective discoveries is crucial to understand the variation in the integrated anomalies that together define craniosynostosis conditions. These associated anomalies contribute to additional health issues critical to effective clinical care of people with craniosynostosis conditions including type and timing of surgery, treatment of comorbidities, and long-term effects on neuropsychological aspects and quality of life. Future research applied to large molecular datasets, analysis of pathways and networks, and the complexity of the craniosynostosis phenotype will require integrative analyses by multidisciplinary teams of physicians and scientists including system and developmental biologists, quantitative anatomists, epidemiologists, geneticists, medical specialists, and surgeons.
Abbreviations
- NSC:
-
Nonsyndromic craniosynostosis
- 3D-CT:
-
Three-dimensional computed tomography
- FGF/FGFR:
-
Fibroblast growth factor/fibroblast growth factor receptor
- BMP:
-
Bone morphogenetic protein
- BBS:
-
Bardet–Biedl syndrome
- ERF:
-
ETS2 repressor factor
- TCF12:
-
Transcription factor 12
References
Papers of particular interest, published recently, have been highlighted as: ∙ Of importance ∙∙ Of major importance
Cohen MM, MacLean RE. Craniosynostosis: diagnosis, evaluation, and management. 2nd ed. New York: Oxford University Press; 2000.
Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989–2003. Am J Med Genet A. 2008;146A:984–91.
Hunter AG, Rudd NL. Craniosynostosis. I. Sagittal synostosis: its genetics and associated clinical findings in 214 patients who lacked involvement of the coronal suture(s). Teratology. 1976;14:185–93.
Lajeunie E, Le Merrer M, Marchac D, et al. Syndromal and nonsyndromal primary trigonocephaly: analysis of a series of 237 patients. Am J Med Genet. 1998;75:211–5.
Singer S, Bower C, Southall P, et al. Craniosynostosis in Western Australia, 1980–1994: a population-based study. Am J Med Genet. 1999;83:382–7.
Winter R, Baraitser M. London Dysmorphology Database [online]. http://www.lmdatabases.com. Accessed 22 May 2014.
McKusick VA, Hamosh A. OMIM [online]. http://www.omim.org/. Accessed 22 May 2014.
Lajeunie E, Le Merrer M, Bonaiti-Pellie C, et al. Genetic study of scaphocephaly. Am J Med Genet. 1996;62:282–5.
Heuze Y, Boyadjiev SA, Marsh JL, et al. New insights into the relationship between suture closure and craniofacial dysmorphology in sagittal nonsyndromic craniosynostosis. J Anat. 2010;217:85–96.
Hunter AG, Rudd NL. Craniosynostosis. II. Coronal synostosis: its familial characteristics and associated clinical findings in 109 patients lacking bilateral polysyndactyly or syndactyly. Teratology. 1977;15:301–9.
Lajeunie E, Le Merrer M, Bonaiti-Pellie C, et al. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet. 1995;55:500–4.
Currarino G. Premature closure of the frontozygomatic suture: unusual frontoorbital dysplasia mimicking unilateral coronal synostosis. Am J Neuroradiol. 1985;6:643–6.
Rogers GF, Proctor MR, Mulliken JB. Unilateral fusion of the frontosphenoidal suture: a rare cause of synostotic frontal plagiocephaly. Plast Reconstr Surg. 2002;110:1011–21.
Di Rocco F, Arnaud E, Renier D. Evolution in the frequency of nonsyndromic craniosynostosis. J Neurosurg Pediatr. 2009;4:21–5.
Kolar JC. An epidemiological study of nonsyndromal craniosynostoses. J Craniofac Surg. 2011;22:47–9.
Lajeunie E, Le Merrer M, Arnaud E, Marchac D, Renier D. Trigonocephaly: isolated, associated and syndromic forms. Genetic study in a series of 278 patients. Arch Pediatr. 1998;5:873–9.
Passos-Bueno MR, Serti Eacute AE, Jehee FS, et al. Genetics of craniosynostosis: genes, syndromes, mutations and genotype-phenotype correlations. Front Oral Biol. 2008;12:107–43.
Wilkie AO, Byren JC, Hurst JA, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126:e391–400.
Jabs EW, Lewanda AF. Craniosynostosis. In: Rimoin DL, Pyeritz RE, Korf BR, editors. Emery & Rimoin’s principles and practice of medical genetics. 6th ed. London: Elsevier; 2013. p. 1–34.
∙ Heuze Y, Martinez-Abadias N, Stella JM, et al. Quantification of facial skeletal shape variation in fibroblast growth factor receptor-related craniosynostosis syndromes. Birth Defects Res A Clin Mol Teratol. 2014;100(4):250–9. This is the first quantitative study of facial shape associated with specific FGFR2 mutations in Apert syndrome.
Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207:637–53.
Moloney DM, Wall SA, Ashworth GJ, et al. Prevalence of Pro250Arg mutation of fibroblast growth factor receptor 3 in coronal craniosynostosis. Lancet. 1997;349:1059–62.
Muenke M, Gripp KW, McDonald-McGinn DM, et al. A unique point mutation in the fibroblast growth factor receptor 3 gene (FGFR3) defines a new craniosynostosis syndrome. Am J Hum Genet. 1997;60:555–64.
Doherty ES, Lacbawan F, Hadley DW, et al. Muenke syndrome (FGFR3-related craniosynostosis): expansion of the phenotype and review of the literature. Am J Med Genet A. 2007;143A:3204–15.
Mulliken JB, Gripp KW, Stolle CA, et al. Molecular analysis of patients with synostotic frontal plagiocephaly (unilateral coronal synostosis). Plast Reconstr Surg. 2004;113:1899–909.
Renier D, El-Ghouzzi V, Bonaventure J, et al. Fibroblast growth factor receptor 3 mutation in nonsyndromic coronal synostosis: clinical spectrum, prevalence, and surgical outcome. J Neurosurg. 2000;92:631–6.
Boyadjiev SA. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res. 2007;10:129–37.
Kim S-D, Liu JL, Roscioli T, et al. Leucine-rich repeat, immunoglobulin-like and transmembrane domain 3 (LRIT3) is a modulator of FGFR1. FEBS Lett. 2012;586:1516–21.
Kress W, Schropp C, Lieb G, et al. Saethre–Chotzen syndrome caused by TWIST 1 gene mutations: functional differentiation from Muenke coronal synostosis syndrome. Eur J Hum Genet. 2006;14:39–48.
Seto ML, Hing AV, Chang J, et al. Isolated sagittal and coronal craniosynostosis associated with TWIST box mutations. Am J Med Genet A. 2007;143:678–86.
Yagnik G, Ghuman A, Kim S, et al. ALX4 gain-of-function mutations in nonsyndromic craniosynostosis. Hum Mutat. 2012;33:1626–9.
Merrill AE, Bochukova EG, Brugger SM, et al. Cell mixing at a neural crest-mesoderm boundary and deficient ephrin–Eph signaling in the pathogenesis of craniosynostosis. Hum Mol Genet. 2006;15:1319–28.
Weber I, Ninkovic M, Janicke A, et al. Molecular analysis of 74 patients with craniosynostosis. Eur J Hum Genet. 2001;9:P0409.
Cunningham ML, Horst JA, Rieder MJ, et al. IGF1R variants associated with isolated single suture craniosynostosis. Am J Med Genet A. 2011;155A:91–7.
Johnson D, Wall SA, Mann S, et al. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? Eur J Hum Genet. 2000;8:571–7.
Kress W, Petersen B, Collmann H, et al. An unusual FGFR1 mutation (fibroblast growth factor receptor 1 mutation) in a girl with non-syndromic trigonocephaly. Cytogenet Cell Genet. 2000;91:138–40.
Mefford HC, Shafer N, Antonacci F, et al. Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynostosis. Am J Med Genet A. 2010;152A:2203–10.
Vissers LE, Cox TC, Maga AM, et al. Heterozygous mutations of FREM1 are associated with an increased risk of isolated metopic craniosynostosis in humans and mice. PLoS Genet. 2011;7:e1002278.
∙∙ Justice CM, Yagnik G, Kim Y, et al. A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat Genet. 2012;44:1360–1364. This study is the first genome wide association study on nonsyndromic craniosynostosis, showing strong evidence for two new genetic loci.
∙∙ Twigg SR, Vorgia E, McGowan SJ, et al. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet. 2013;45:308–313. This study uses whole exome sequencing to identify a new gene, involved in the MAPK pathway, with mutations in patients with nonsyndromic sagittal or lambdoid synostosis and other craniosynostoses.
∙∙ Sharma VP, Fenwick AL, Brockop MS, et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet. 2013;45:304–307. This study uses whole exome sequencing to identify a new gene with mutations for syndromic and nonsyndromic coronal synostosis.
Rosen V. BMP2 signaling in bone development and repair. Cytokine Growth Factor Rev. 2009;20:475–80.
Connerney J, Andreeva V, Leshem Y, et al. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol. 2008;318:323–34.
Marie PJ, Debiais F, Hay E. Regulation of human cranial osteoblast phenotype by FGF-2, FGFR-2 and BMP-2 signaling. Histol Histopathol. 2002;17:877–85.
Maxson R, Ishii M. The Bmp pathway in skull vault development. Front Oral Biol. 2008;12:197–208.
Jin H, Nachury MV. The BBSome. Curr Biol. 2009;19:R472–3.
Nachury MV, Loktev AV, Zhang Q, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129:1201–13.
Pedersen LB, Rosenbaum JL. Intraflagellar transport (IFT) role in ciliary assembly, resorption and signalling. Curr Top Dev Biol. 2008;85:23–61.
Tobin JL, Beales PL. Bardet–Biedl syndrome: beyond the cilium. Pediatr Nephrol. 2007;22:926–36.
Walczak-Sztulpa J, Eggenschwiler J, Osborn D, et al. Cranioectodermal Dysplasia, Sensenbrenner syndrome, is a ciliopathy caused by mutations in the IFT122 gene. Am J Hum Genet. 2010;86:949–56.
Arts HH, Bongers EM, Mans DA, et al. C14ORF179 encoding IFT43 is mutated in Sensenbrenner syndrome. J Med Genet. 2011;48:390–5.
Bredrup C, Saunier S, Oud MM, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89:634–43.
Gilissen C, Arts HH, Hoischen A, et al. Exome sequencing identifies WDR35 variants involved in Sensenbrenner syndrome. Am J Hum Genet. 2010;87:418–23.
Schmelzer RE, Perlyn CA, Kane AA, et al. Identifying reproducible patterns of calvarial dysmorphology in nonsyndromic sagittal craniosynostosis may affect operative intervention and outcomes assessment. Plast Reconstr Surg. 2007;119:1546–52.
Johnson D, Wilkie AO. Craniosynostosis. Eur J Hum Genet. 2011;19:369–76.
Le Tanno P, Poreau B, Devillard F, et al. Maternal complex chromosomal rearrangement leads to TCF12 microdeletion in a patient presenting with coronal craniosynostosis and intellectual disability. Am J Med Genet A. 2014;164(6):1530–6.
el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, et al. Mutations of the TWIST gene in the Saethre–Chotzen syndrome. Nat Genet. 1997;15:42–6.
Howard TD, Paznekas WA, Green ED, et al. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre–Chotzen syndrome. Nat Genet. 1997;15:36–41.
Chai Y, Maxson RE. Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–75.
Jabs EW. TWIST and the Saethre–Chotzen syndrome. In: Epstein CJ, Erickson R, Wynshaw-Boris A, editors. Inborn Erros Dev Mol Basis Clin Disord Morphog. Oxford: Oxford University Press; 2008. p. 474–81.
Bernardini C, Barba M, Tamburrini G, et al. Gene expression profiling in human craniosynostoses: a tool to investigate the molecular basis of suture ossification. Childs Nerv Syst. 2012;28:1295–300.
Stamper BD, Park SS, Beyer RP, et al. Differential expression of extracellular matrix-mediated pathways in single-suture craniosynostosis. PLoS One. 2011;6:e26557.
∙ Holmes G. The role of vertebrate models in understanding craniosynostosis. Childs Nerv Syst. 2012;(28):1471–1481. This is a review article highlighting progress in mouse models for craniosynostosis.
Holmes G. Mouse models of Apert syndrome. Childs Nerv Syst. 2012;28:1505–10.
Johnson D, Iseki S, Wilkie AO, et al. Expression patterns of Twist and Fgfr1, -2 and -3 in the developing mouse coronal suture suggest a key role for twist in suture initiation and biogenesis. Mech Dev. 2000;91:341–5.
Rice DP, Connor EC, Veltmaat JM, et al. Gli3Xt-J/Xt-J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum Mol Genet. 2010;19:3457–67.
Perlyn C, Morriss-Kay G, Darvann T, et al. A model for the pharmacological treatment of crouzon syndrome. Neurosurgery. 2006;59:210–5; discussion 210–215.
Shukla V, Coumoul X, Wang R, et al. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet. 2007;39:1145–50.
Yin L, Du X, Li C, et al. A Pro253Arg mutation in fibroblast growth factor receptor 2 (Fgfr2) causes skeleton malformation mimicking human Apert syndrome by affecting both chondrogenesis and osteogenesis. Bone. 2008;42:631–43.
Wang Y, Zhou X, Oberoi K, et al. p38 Inhibition ameliorates skin and skull abnormalities in Fgfr2 Beare-Stevenson mice. J Clin Invest. 2012;122:2153–64.
∙ Motch Perrine SM, Cole TM, 3rd, Martinez-Abadias N, et al. Craniofacial divergence by distinct prenatal growth patterns in Fgfr2 mutant mice. BMC Dev Biol. 2014;14:8. This study provides evidence of the differential contribution of different craniosynostosis causing mutations to craniofacial dysmorphogenesis.
∙ Percival CJ, Huang Y, Jabs EW, et al. Embryonic craniofacial bone volume and bone mineral density in Fgfr2(+/P253R) and nonmutant mice. Dev Dyn. 2014;243:541-551. This study presents quantification of the typical pattern of cranial bone growth and maturation during early osteogenesis and as modified in by craniosynostosis causing mutations in mice.
Wang Y, Sun M, Uhlhorn VL, et al. Activation of p38 MAPK pathway in the skull abnormalities of Apert syndrome Fgfr2(+P253R) mice. BMC Dev Biol. 2010;10:22.
Wang Y, Xiao R, Yang F, et al. Abnormalities in cartilage and bone development in the Apert syndrome FGFR2(+/S252W) mouse. Development. 2005;132:3537–48.
Hill CA, Martinez-Abadias N, Motch SM, et al. Postnatal brain and skull growth in an Apert syndrome mouse model. Am J Med Genet A. 2013;161A:745–57.
∙ Martinez-Abadias N, Motch SM, Pankratz TL, et al. Tissue-specific responses to aberrant FGF signaling in complex head phenotypes. Dev Dyn. 2013;242:80–94. This study provides evidence of the effect of craniosynostosis causing mutations on cranial soft tissues and their secondary effect of bone dysmorphogenesis.
Heuze Y, Singh N, Basilico C, et al. Morphological comparison of the craniofacial phenotypes of mouse models expressing the Apert FGFR2 S252W mutation in neural crest- or mesoderm-derived tissues. Bone. 2014;63:101–9.
Sharpe J, Ahlgren U, Perry P, et al. Optical projection tomography as a tool for 3D microscopy and gene expression studies. Science. 2002;296:541–5.
Hiraki Y, Miyatake S, Hayashidani M, et al. Aortic aneurysm and craniosynostosis in a family with Cantu syndrome. Am J Med Genet A. 2014;164A:231–6.
Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007;2:40.
Weiss MJ, Cole DE, Ray K, et al. A missense mutation in the human liver/bone/kidney alkaline phosphatase gene causing a lethal form of hypophosphatasia. Proc Natl Acad Sci USA. 1988;85:7666–9.
Kaname T, Yanagi K, Chinen Y, et al. Mutations in CD96, a member of the immunoglobulin superfamily, cause a form of the C (Opitz trigonocephaly) syndrome. Am J Hum Genet. 2007;81:835–41.
Searle C, Jewell R, Kraft J, et al. Craniosynostosis: a previously unreported association with CHST3-related skeletal dysplasia (autosomal recessive Larsen syndrome). Clin Dysmorphol. 2014;23:12–5.
Laue K, Pogoda HM, Daniel PB, et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am J Hum Genet. 2011;89:595–606.
Twigg SR, Kan R, Babbs C, et al. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci USA. 2004;101:8652–7.
Simpson MA, Scheuerle A, Hurst J, et al. Mutations in FAM20C also identified in non-lethal osteosclerotic bone dysplasia. Clin Genet. 2009;75:271–6.
Kosaki K, Takahashi D, Udaka T, et al. Molecular pathology of Shprintzen–Goldberg syndrome. Am J Med Genet A. 2006;140:104–108; author reply 109–110.
Jehee FS, Bertola DR, Yelavarthi KK, et al. An 11q11–q13.3 duplication, including FGF3 and FGF4 genes, in a patient with syndromic multiple craniosynostoses. Am J Med Genet A. 2007;143A:1912–1918.
White KE, Cabral JM, Davis SI, et al. Mutations that cause osteoglophonic dysplasia define novel roles for FGFR1 in bone elongation. Am J Hum Genet. 2005;76:361–7.
Muenke M, Schell U, Hehr A, et al. A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat Genet. 1994;8:269–74.
Chun K, Siegel-Bartelt J, Chitayat D, et al. FGFR2 mutation associated with clinical manifestations consistent with Antley–Bixler syndrome. Am J Med Genet. 1998;77:219–24.
Wilkie AO, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–72.
Park WJ, Theda C, Maestri NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet. 1995;57:321–8.
Przylepa KA, Paznekas W, Zhang M, et al. Fibroblast growth factor receptor 2 mutations in Beare–Stevenson cutis gyrata syndrome. Nat Genet. 1996;13:492–4.
Merrill AE, Sarukhanov A, Krejci P, et al. Bent bone dysplasia-FGFR2 type, a distinct skeletal disorder, has deficient canonical FGF signaling. Am J Hum Genet. 2012;90:550–7.
Reardon W, Winter RM, Rutland P, et al. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 1994;8:98–103.
Jabs EW, Li X, Scott AF, et al. Jackson–Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8:275–9.
Lajeunie E, Ma HW, Bonaventure J, et al. FGFR2 mutations in Pfeiffer syndrome. Nat Genet. 1995;9:108.
Schell U, Hehr A, Feldman GJ, et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer syndrome. Hum Mol Genet. 1995;4:323–8.
Rutland P, Pulleyn LJ, Reardon W, et al. Identical mutations in the FGFR2 gene cause both Pfeiffer and Crouzon syndrome phenotypes. Nat Genet. 1995;9:173–6.
Kan SH, Elanko N, Johnson D, et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am J Hum Genet. 2002;70:472–86.
Meyers GA, Orlow SJ, Munro IR, et al. Fibroblast growth factor receptor 3 (FGFR3) transmembrane mutation in Crouzon syndrome with acanthosis nigricans. Nat Genet. 1995;11:462–4.
Lajeunie E, El Ghouzzi V, Le Merrer M, et al. Sex related expressivity of the phenotype in coronal craniosynostosis caused by the recurrent P250R FGFR3 mutation. J Med Genet. 1999;36:9–13.
Tavormina PL, Shiang R, Thompson LM, et al. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat Genet. 1995;9:321–8.
Hurst JA, Jenkins D, Vasudevan PC, et al. Metopic and sagittal synostosis in Greig cephalopolysyndactyly syndrome: five cases with intragenic mutations or complete deletions of GLI3. Eur J Hum Genet. 2011;19:757–62.
McDonald-McGinn DM, Feret H, Nah HD, et al. Metopic craniosynostosis due to mutations in GLI3: a novel association. Am J Med Genet A. 2010;152A:1654–60.
Villarreal DD, Villarreal H, Paez AM, et al. A patient with a unique frameshift mutation in GPC3, causing Simpson–Golabi–Behmel syndrome, presenting with craniosynostosis, penoscrotal hypospadias, and a large prostatic utricle. Am J Med Genet A. 2013;161A:3121–5.
Klopocki E, Lohan S, Brancati F, et al. Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am J Hum Genet. 2011;88:70–5.
Nieminen P, Morgan NV, Fenwick AL, et al. Inactivation of IL11 signaling causes craniosynostosis, delayed tooth eruption, and supernumerary teeth. Am J Hum Genet. 2011;89:67–81.
Yilmaz S, Turhan T, Mutluer S, et al. The association of Alagille syndrome and craniosynostosis. Pediatr Neurol. 2013;48:146–8.
Kamath BM, Stolle C, Bason L, et al. Craniosynostosis in Alagille syndrome. Am J Med Genet. 2002;112:176–80.
Twigg SR, Lloyd D, Jenkins D, et al. Mutations in multidomain protein MEGF8 identify a Carpenter syndrome subtype associated with defective lateralization. Am J Hum Genet. 2012;91:897–905.
Jabs EW, Muller U, Li X, et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75:443–50.
Janssen A, Hosen MJ, Jeannin P, et al. Second family with the Boston-type craniosynostosis syndrome: novel mutation and expansion of the clinical spectrum. Am J Med Genet A. 2013;161:2352–7.
Florisson JM, Verkerk AJ, Huigh D, et al. Boston type craniosynostosis: report of a second mutation in MSX2. Am J Med Genet Part A. 2013;161:2626–33.
Bernardini L, Castori M, Capalbo A, et al. Syndromic craniosynostosis due to complex chromosome 5 rearrangement and MSX2 gene triplication. Am J Med Genet A. 2007;143A:2937–43.
Shiihara T, Kato M, Kimura T, et al. Craniosynostosis with extra copy of MSX2 in a patient with partial 5q-trisomy. Am J Med Genet A. 2004;128A:214–6.
Mahmoud Adel AH, Abdullah AA, Eissa F. Infantile osteopetrosis, craniosynostosis, and Chiari malformation type I with novel OSTEM1 mutation. J Pediatr Neurosci. 2013;8:34–7.
Huang N, Pandey AV, Agrawal V, et al. Diversity and function of mutations in p450 oxidoreductase in patients with Antley–Bixler syndrome and disordered steroidogenesis. Am J Hun Genet. 2005;76:729–49.
Jenkins D, Seelow D, Jehee FS, et al. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007;80:1162–70.
Van Maldergem L, Siitonen HA, Jalkh N, et al. Revisiting the craniosynostosis-radial ray hypoplasia association: Baller-Gerold syndrome caused by mutations in the RECQL4 gene. J Med Genet. 2006;43:148–52.
Wilson GR, Sunley J, Smith KR, et al. Mutations in SH3PXD2B cause Borrone dermato-cardio-skeletal syndrome. Eur J Hum Genet. 2013;22(6):741–7.
Bendon CL, Fenwick AL, Hurst JA, et al. Frank-ter Haar syndrome associated with sagittal craniosynostosis and raised intracranial pressure. BMC Med Genet. 2012;13:104.
Carmignac V, Thevenon J, Ades L, et al. In-frame mutations in exon 1 of SKI cause dominant Shprintzen–Goldberg syndrome. Am J Hum Genet. 2012;91:950–7.
Tagariello A, Heller R, Greven A, et al. Balanced translocation in a patient with craniosynostosis disrupts the SOX6 gene and an evolutionarily conserved non-transcribed region. J Med Genet. 2006;43:534–40.
Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–81.
Van Hemelrijk C, Renard M, Loeys B. The Loeys–Dietz syndrome: an update for the clinician. Curr Opin Cardiol. 2010;25:546–51.
Hartill VL, Pendlebury M, Hobson E. Mowat–Wilson syndrome associated with craniosynostosis. Clin Dysmorphol. 2014;23:16–9.
Acknowledgments
The work was supported in part by Grants NIH/NIDCR 1 R01 DE022988, R01 DE018500, and 3 R01 DE18500-02S1.
Disclosure
Y Heuzé, G Holmes, I Peter, JT Richtsmeier, and EW Jabs all declare no conflicts of interest.
Human and Animal Rights and Informed Consent
All studies by Y Heuzé, G Holmes, I Peter, JT Richtsmeier, and EW Jabs involving animal and/or human subjects were performed after the approval by the appropriate institutional review boards. When required, written informed consent was obtained from all participants.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Heuzé, Y., Holmes, G., Peter, I. et al. Closing the Gap: Genetic and Genomic Continuum from Syndromic to Nonsyndromic Craniosynostoses. Curr Genet Med Rep 2, 135–145 (2014). https://doi.org/10.1007/s40142-014-0042-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-014-0042-x