
Abstract
Three new pyridine type alkaloids, (−)-vinmajpyridines A–C (1–3), along with two known alkaloids, have been isolated from the aerial parts of Vinca major cultivated in Pakistan. Their structures have been elucidated by means of NMR and HRESIMS spectroscopic data. The new alkaloids were evaluated for their cytotoxicity against glioma initiating cell lines (GITC-3# and GITC-18#), glioblastoma cell lines (U-87MG and T98G), and lung cancer cell line A-549, but none of them was active at 20 μg/mL concentration.
Similar content being viewed by others
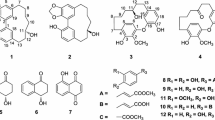


1 Introduction
The genus Vinca (Apocynaceae), distributed through Europe, Northwest Africa, and South-west Asia, represents a group of species which are rich in indole alkaloids of diverse structural patterns, many of which are of considerable therapeutic value [1,2,3,4]. Vinca major has been used for centuries as a folk remedy in the treatment of menorrhagia and diabetes, and as an abortifacient and vulnerary [5]. Delphinidin glycosides have been isolated from the flowers of V. major [6] while chlorogenic acid, robinin, and flavonol triglycoside are extracted from its leaves [7]. In addition, a number of indole alkaloids are also reported from this plant [8,9,10,11,12,13]. Previously, we isolated non-alkaloid constituents [14] as well as indole alkaloids [15] from V. major cultivated in Kunming. In our ongoing search for alkaloids from this plant growing in different habitats, we carried out the phytochemical investigation of the aerial parts of V. major in Pakistan, and then isolated three new pyridine alkaloids, named as (–)-vincapyridines A–C (1–3) trivially (Fig. 1). The structures of new alkaloids were elucidated by means of spectroscopic methods. The new alkaloids were evaluated for their cytotoxicity against human glioma initiating cell lines (GITC-3# and GITC-18#), glioblastoma cell lines (U-87MG and T98G), and lung cancer cell line A-549 using the reported MTS assay with DMSO as the control group. Unfortunately, none of these compounds exhibited significant cytotoxicity at 20 μg/mL concentration. Herein, we report the isolation, structural elucidation of these compounds.

Structures of compounds 1–5
2 Results and Discussion
The molecular formula of 1 was determined to be C13H17NO4, by a quasi-molecular ion peak at m/z 252.1222 [M+H]+ (calcd for C13H18NO4, 252.1230) in the positive HRESIMS. Its IR spectrum revealed a characteristic absorption band at 1724 (C=O). The 1H and 13C NMR spectral data of 1 (Table 1), assumed that 1 was a tri-substituents pyridine derivative with two typical downfield protons [δ H 8.89 (s) and 8.53 (s)] assigned to be pyridine aromatic protons [16]. In HMBC spectrum of 1, correlations of pyridine protons δ C 8.53 (s) with substituents aromatic carbons at δ C 146.3 (C-7), 137.6 (C-2), and oxymethine at δ C 71.3 (C-3), indicated substituents pattern (Fig. 2). Moreover, HMBC correlations of δ H 4.88 (H-5) with δ C 71.3 (C-3) and δ C 146.3 (C-7) established a substituents pyranose ring fused to the pyridine ring (Fig. 2). Besides, HMBC correlations of δ H 1.64 (3H, d, –CH3) with δ C 71.3 (C-3), and of δ H 3.99 and 3.62 (2H, –OCH 2CH3) with δ C 99.6 (C-5), placed a methyl at C-6 and oxyethyl group at C-5, respectively, which was further supported by the correlations of δ H 3.37/4.88, 3.99/1.24, and 5.02/1.64, in its 1H–1H COSY spectrum (Fig. 2). Finally, the HMBC correlations of δ H 8.89 (s) and 3.92 (–OCH 3) with δ C 167.2 (–COOCH3) positioned the methyl formate group at C-8. In ROESY spectrum of 1, NOE correlation between δ H 4.88 (H-5) and 1.64 (H-4) revealed two protons to be co-facial (Fig. 2), which indicated its relative configuration.

Selective HMBC (→), 1H–1H COSY (−), and ROESY (↔) correlations for 1–3
The molecular formula of 2 was established as C12H15NO4 by a quasi-molecular ion peak in the HRESIMS at m/z 238.1072 [M+H]+ (calcd for C12H16NO4, 238.1074). The 1H and 13C NMR spectral data of 2 were similar to those of 1, except for a methoxyl group connected to C-5 in 2 instead of the ethoxyl group in 1, which was consistent with its molecular formula, and further supported by HMBC correlation. Compound 2 shared same relative configurations with 1, supported by NOE correlation between H-5 and H-4 (Fig. 2).
Its molecular formula of 3 was deduced to be C11H15NO2 on the basis of HRESIMS quasi-molecular ion peak at m/z 194.1172 [M+H]+ (calcd for C11H16NO2, 194.1176) and 13C NMR data (Table 1). Signals for methyl formate group at C-8 in the 1H and 13C NMR spectra of 1 were absent in those of 3,and corresponding δ H 7.18 (d, 5.2, H-8) and δ C 125.5 (d, C-8) were appeared in the 1H and 13C NMR spectra of 3. Other parts of 3 were identical to those of 1, supported by its HMBC and ROSEY spectra (Fig. 2).
The optical rotation of 1–3 with same negative sign, supposed their same absolute configuration. However, the specific rotation values of 1–3 were obviously different, which indicated that substituents at C-8 and 5 contributed to specific rotation significantly. In compound 1, to avoid steric hindrance between –COOCH3 and –OCH2CH3 (Fig. 3), the dihedral angle between H-6 and H-5 were changed, resulting in a large coupling constants (J = 8.4 Hz) for H-6/H-5 in its 1H NMR spectrum, which were different from those of two other compounds. Meanwhile, the deformed substituent pyran forward the hemiacetal proton (H-5) to the shield area of pyridine ring (Fig. 3), which caused the up-field chemical shift of H-5 (δ H 4.88) in 1, comparison of those in 2 (δ H 5.07) and 3 (δ H 5.17). It is the first report of pyridine type alkaloids from genus Vinca. Besides, known indole alkaloids nauclefine (4) [17, 18] and vincamajoreine (5) [19] were also isolated.

Conformation change of compound 1 indicated by molecular model
3 Experimental Section
3.1 Plant Material
The aerial parts of V. major were collected from Oghi, Mansehra, KPK, Pakistan in June, 2015 and identified by one of us (A. Khan). A voucher specimen (No. Khan_20150601) has been deposited in the State Key Laboratory of Phytochemistry and Plant Resources in West China, Kunming Institute of Botany, Chinese Academy of Sciences, China.
3.2 General Experimental Procedures
Optical rotations were performed on a P-1020 polarimeter. IR spectra were measured on a Bruker FT-IR Tensor 27 spectrometer with KBr pellets. UV spectra were obtained on Shimadzu UV-2401A spectrometer. 1D and 2D-NMR spectra were recorded on Bruker AV-600 MHz spectrometer. Coupling constants were expressed in Hertz and chemical shifts were given on a ppm scale with tetramethylsilane as an internal standard. HRESIMS were recorded on an API QSTAR Pulsar 1 spectrometer. Column chromatography (CC) was performed on silica gel (200–300 mesh, Qingdao Marine Chemical Ltd., Qingdao, People’s Republic of China), Sephadex LH-20 (Pharmacia Fine Chemical Co., Ltd., Sweden), and MCI-gel CHP 20P (75–100 μm, Mitsubishi Chemical Co., Ltd). Thin-layer chromatography (TLC) was carried out on pre-coated silica gel plates (Qingdao Marine Chemical Co., Ltd.) with CHCl3/MeOH (15:1, 4:1, v/v) as developing solvents and spots were visualized by Dragendorff’s reagent. High performance liquid chromatography (HPLC) was performed using Waters 600 pump with semi-preparative C18 columns (150 × 9.4).
3.3 Extraction and Isolation
The air-dried and powdered aerial parts of V. major (8 kg) were extracted with 80% aqueous MeOH (80 L × 3) at room temperature. After removal of the organic solvent under reduced pressure, the residue was dissolved in 0.3% aqueous hydrochloric acid (v/v). The solution was subsequently basified to pH 9–10 using aqueous ammonia, and then extracted with EtOAc (3 L × 4) to give an alkaloidal extract (34.3 g). The extract was applied to a silica gel column (CHCl3/MeOH, 1:0–0:1) to afford nine fractions (Fr. A-I). Fr. B (2.5 g) was subjected to silica gel column chromatography (CC) using a petroleum ether/acetone gradient eluent (10:1–9:1) to afford sub-fractions (Fr. 1–5). Fr. 4 (200 mg) was further purified on MCI-gel CHP 20P column using a MeOH/H2O gradient eluent (1:4–1:0) and on a semi-preparative C18 HPLC column with a gradient of MeOH/H2O (50:50–90:10) to yield vincamajoreine (5) (6 mg). Fr. H (5 g) was subjected to silica gel CC (CHCl3/MeOH, 9:1–0:1) to afford sub-fractions (Fr. 5–10). Fr. 5 (1200 mg) was further separated on MCI-gel CHP 20P column to yield nauclefine (4) (15 mg). Fr. 8 (800 mg) was subjected to Sephadex LH-20 CC using MeOH under isocratic conditions and was further separated on a semi-preparative C18 HPLC column with a gradient of MeOH/H2O (30:70–70:30) to produce (−)-vincapyridine A (1) (3 mg), (−)-vincapyridine B (2) (2 mg), and (−)-vincapyridine C (3) (1.2 mg).
(−)-vincapyridine A (1): C13H17NO4. white amorphous powder; \(\left[ {\upalpha } \right]_{\text{D}}^{26}\) −26.6 (c 0.07, MeOH); UV (MeOH) λ max (log ε): 203 (4.22), 220 (3.92), 270 (3.38); IR (KBr) ν max 3418, 2929, 1724, 1572, 1381, 1295, 1079 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 252.1222 [M+H]+ (calcd for C13H18NO4, 252.1230).
(−)-vincapyridine B (2): C12H15NO4. white amorphous powder; \(\left[ {\upalpha } \right]_{\text{D}}^{26}\) −190.0 (c 0.04, MeOH); UV (MeOH) λ max (log ε): 204 (4.44), 219 (4.13), 269 (3.57); IR (KBr) ν max 3419, 2926, 1724, 1606, 1383, 1042 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 238.1072 [M+H]+ (calcd for C12H16NO4, 238.1074).
(−)-vincapyridine C (3): C11H15NO2. white amorphous powder; \(\left[ {\upalpha } \right]_{\text{D}}^{26}\) −359.8 (c 0.02, MeOH); UV (MeOH) λ max (log ε): 204 (3.35), 279 (2.64); IR (KBr) ν max 3425, 2926, 1720, 1623, 1383, 1035 cm−1; 1H and 13C NMR data, see Table 1; HRESIMS m/z 194.1172 [M+H]+ (calcd for C11H16NO2, 194.1176).
3.4 Cytotoxicity Assay
GITC-3# and GITC-18# (glioma initiating cell lines) were established previously in Kunming Institute of Zoology from three different human glioblastoma multiform samples. These cell lines were cultured in serum-free medium supplemented with 1XB27 (Life 12587-010), 50 ng/mL EGF (PeproTech AF-100-15), and bFGF (PeproTech AF-100-18B). GITCs were cultured in laminin (Gibco 1725712) pre-mdish. The cells could adhere and normally grow without differentiation. Culture dishes were pre-coated with laminin for 4-6 h at 10 mg/mL concentration. The T98G, U-87MG, and A549 cell lines were purchased from the American Type Culture Collection (ATCC), the cells were cultured in DMEM basic medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified incubator at 37 °C and an atmosphere of 5% CO2. Cells were digested with TryplE express (Gibco 12604-021) for 3–5 min at 37 °C in cell incubator and centrifuged at 1000 rpm/min for 3 min.
Cell viability analysis was performed by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, Promega #G3581] assay [20, 21]. The cells were digested and seeded on a 96-well plate with 20000 cells/well. Each tumor cell line was exposed to the test compound dissolved in DMSO at concentrations of 20 μg/mL and kept in cell incubator for 72 h. MTS reagent was diluted 1:5 with fresh medium and mixed well. The old medium was removed and subsequently the fresh medium was added with 100 μL/well. The cells were incubated for 1 h. Absorbance was measured by Hybrid Reader (BioTek Synergy H1) at 490 nm. The cell viability was evaluated by percentage compared with DMSO as a control group.
4 Supplementary Information
1D and 2D NMR spectra, HRESIMS, and UV spectra of compounds 1–3 are available as Supplementary Information.
References
J. Balsevich, F. Constabel, W.G.W. Kurz, Planta Med. 44, 91–93 (1982)
A. Banerji, M. Chakrabarty, Phytochemistry 13, 2309–2312 (1974)
A. Banerji, M. Chakrabarty, Phytochemistry 16, 1124–1125 (1977)
A. Chatterjee, A. Banerji, M. Chakrabarty, Planta Med. 28, 109–111 (1975)
N.R. Farnsworth, H.H.S. Fong, R.N. Blomster, F.J. Draus, J. Pharm. Sci. 51, 217–224 (1962)
N. Ishikura, K. Minekishi, Bot. Mag. Tokyo. 91, 181–186 (1978)
A. Sakushima, S. Nishibe, Phytochemistry 27, 915–919 (1988)
A.U. Rahman, A. Sultana, F. Nighat, M.K. Bhatti, S. Kurucu, M. Kartal, Phytochemistry 38, 1057–1061 (1995)
F. Bahadori, G. Topcu, M. Boga, A. Turkekul, U. Kolak, M. Kartal, Nat. Prod. Commun. 7, 731–734 (2012)
G. Mukhopadhyay, B. Mukherjee, A. Patra, A. Chatterjee, R. Ghosh, P. Roychowdhury, H. Kawazura, Phytochemistry 30, 2447–2449 (1991)
D. Sohretoglu, M. Masullo, S. Piacente, H. Kirmizibekmez, Biochem. Syst. Ecol. 49, 69–72 (2013)
Z.J. Zhang, R.N. Du, J. He, X.D. Wu, Y. Li, R.T. Li, Q.S. Zhao, J. Asian Nat. Prod. Res. 18, 328–333 (2016)
Z.J. Zhang, J. Yang, J. He, X.D. Wu, L.D. Shao, Y. Li, S.X. Huang, R.T. Li, Q.S. Zhao, Tetrahedron Lett. 55, 6490–6494 (2014)
G.G. Cheng, H.Y. Zhao, L. Liu, Y.L. Zhao, C.W. Song, J. Gu, W.B. Sun, Y.P. Liu, X.D. Luo, Chin. J. Nat. Med. 14, 56–60 (2016)
G.G. Cheng, Y.L. Zhao, Y. Zhang, P.K. Lunga, D.B. Hu, Y. Li, J. Gu, C.W. Song, W.B. Sun, Y.P. Liu, X.D. Luo, Tetrahedron 70, 8723–8729 (2014)
R. Benkrief, A.L. Skaltsounis, F. Tillequin, M. Koch, J. Preset, J. Nat. Prod. 54, 532–541 (1991)
M. Sainsbury, N.L. Uttley, J. Chem. Soc., Perkin Trans 1, 2416–2418 (1976)
M. Sainsbury, B. Webb, Phytochemistry 14, 2691–2693 (1975)
M.M. Janot, M.J. Le, Ann. Pharm. Fr. 13, 325–328 (1955)
Z. Dai, S.R. Li, P.F. Zhu, L. Liu, B. Wang, Y.P. Liu, X.D. Luo, X.D. Zhao, Bioorg. Med. Chem. Lett. 27, 2863–2867 (2017)
B. Wang, Z. Dai, L. Liu, X. Wei, P.F. Zhu, H.F. Yu, Y.P. Liu, X.D. Luo, Nat. Prod. Bioprosp. 6, 1–6 (2016)
Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (81225024) for financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Xin Wei and Afsar Khan have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wei, X., Khan, A., Song, D. et al. Three New Pyridine Alkaloids from Vinca major Cultivated in Pakistan. Nat. Prod. Bioprospect. 7, 323–327 (2017). https://doi.org/10.1007/s13659-017-0137-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13659-017-0137-7