
Abstract
Key message
Past demographic changes and current selection pressures determine the genetic variation displayed by Nothofagus species along rainfall gradients. Based on the diversity trends observed at candidate genes associated to drought stress, we inferred a differential species’ adaptive potential.
Context
Clinal genetic variation in natural populations could reflect either recent demographic history or the evolution of adapted genotypes along heterogeneous environments.
Aims
We describe genetic variation patterns in three Nothofagus species of South American temperate forests, growing along steep rainfall gradients. Our hypothesis is that the selection pressure along this gradient reinforces the genetic structure previously shaped by Pleistocene climate oscillations.
Methods
We screened variation along gradients at putative adaptive markers: candidate genes involved in response to drought, and EST-SSRs linked to drought stress genes. Genomic SSRs (gSSRs) were used to decouple the incidence of demographic events in the genetic structure.
Results
Genetic diversity at SSRs agreed with the putative location of cryptic Pleistocene refugia in Nothofagus. In addition, each species showed different trends for nucleotide diversity at candidate genes. Unbiased heterozygosity significantly correlated with precipitation at EST-SSRs in Nothofagus nervosa. We found evidences of balancing selection and several SNPs departed from neutral expectations.
Conclusions
Nothofagus genetic variability shows a strong imprint of demographic changes that reveals refugia location for the species during Pleistocene. This variability is modelled by environmental conditions across natural gradients, which impose selection pressure at genome regions related to stress response, providing clues about inter-specific differences in adaptive potential to water deficit.
Similar content being viewed by others

1 Introduction
Climate is currently experiencing significant changes on a global scale, thus affecting ecosystems and generating shifting environments (Allen et al. 2010). The impact on tree species is of concern due to their role as keystone species in the structuring of forests and maintenance of biodiversity (Kremer et al. 2012). In this context, species survival is challenging and there are different possible responses: either be sufficiently plastic, migrate to more suitable areas, rely on genetic adaptation, or become extinct (Aitken et al. 2008; Pardo-Diaz et al. 2015). The ability to adapt to novel conditions will ultimately depend on the intensity of change throughout species ranges and the level of genetic diversity within populations (Mosca et al. 2012; Alberto et al. 2013; Fady et al. 2016). To deal with selective pressures, trees should respond through differential gene expression or exposing a high number of genetic variants to selection (Savolainen et al. 2011). However, the need for adaptation to current rapid climate changes probably exceeds what long-lived organisms like trees may be able to cope with (Fady et al. 2016; Roschanski et al. 2016).
In Patagonia, climatic registers from the last century show that there has been an increase in mean annual temperature (Villalba et al. 2005), as well as a decrease in mean annual precipitation, and general glacial recession (Masiokas et al. 2008). Projections for the future (2020–2040 period), based on the HadCM3 model, predict higher temperatures and decreasing precipitation regimes (Centro de Investigaciones del Mar y de la Atmósfera 2015). These tendencies would turn the area into a more xeric region (Rusticucci and Barrucand 2004; Castañeda and González 2008), with strong seasonality and increasing frequency of extreme climate events (e.g. drought) (Alexander et al. 2006), probably affecting forest growth (e.g. Villalba et al. 2012). Under this situation, the natural environmental gradients along which species occur provide an interesting approach to seek into the genetic diversity trends including adaptive responses. This would be a first step to detect clinal genetic variation along these gradients that could give evidence for the action of selection (Grivet et al. 2011; Chen et al. 2012).
In the North-Patagonian forests, several tree species (including Nothofagus spp.) grow along a marked rainfall gradient imposed by the Andes Mountains´ rain shadow. Accordingly, precipitations decrease from west to east varying with geographical longitude. Along the evolutionary history of many plant species, the concurrent shifts in climate may have influenced the establishment of rainfall gradients in the region, which could have shaped genetic diversity. As reported for herbs, not only temperature (i.e. ice-ages) but also precipitation regimes could have been historically affecting plant species distribution, e.g. Arid Diagonal shifts (Cosacov et al. 2010, 2012). Nothofagus nervosa (= alpina) (Phil) Dim. et Mil. (Raulí), Nothofagus obliqua (Mirb.) Oerst. (Roble Pellín) and Nothofagus pumilio (Poepp. & Endl.) Krasser (Lenga) are three of the most important species growing in North Patagonian forests. All of them possess an outstanding wood quality as well as a relevant role for the structuring of forest ecosystems. Nothofagus nervosa and Nothofagus obliqua are sister species which co-occur along most of their distribution in Argentina, have a common evolutionary history and comparable phylogeographic patterns (Marchelli and Gallo 2006; Azpilicueta et al. 2009). Notwithstanding, these species differ in physiological aspects, such as resistance to drought (Varela et al. 2012), and show differences in their distribution across precipitation gradients. Whereas Nothofagus nervosa inhabits regions from around 1200 up to 3000 mm/year, range limits of Nothofagus obliqua is extended from around 800 up to 3000 mm/year being more tolerant to drier environments. In turn, Nothofagus pumilio, a cold-tolerant species inhabiting the higher altitudinal niche, conform edge and small populations at the easternmost, driest regions of the gradient (steppe-like environment), as well as extended populations at the humid western locations (i.e. from 200 to 2500 mm/year). The described climatic variation is thought to confer strong selective pressure to all the species.
In heterogeneous environments, clines of genetic variation could reflect the evolution of adapted genotypes or recent demographic history (Kawecki and Ebert 2004). At the molecular level, it is difficult to discern how selection affects patterns of nucleotide variation as demographic forces like drift and variation in population size can produce similar patterns (e.g. Heuertz et al. 2006). Gene flow may also be responsible of introducing better adapted variants, thus contributing in shaping adaptive genetic variation (e.g. Pluess et al. 2016). The three Nothofagus species studied here have distinct demographic histories that can have a strong effect on the patterns of variation. Pleistocene glaciations affected the whole region and multiple glacial refugia were proposed, mainly located in western sites for Nothofagus nervosa and Nothofagus pumilio, while Nothofagus obliqua apparently survived to the east (Marchelli et al. 1998; Azpilicueta et al. 2009; Soliani et al. 2015; Marchelli et al. 2017; Mathiasen and Premoli 2010). Interspecific differences in the main ecological traits, like cold and drought tolerance, reinforce these results.
Comparing genetic variation at putatively adaptive vs. neutral markers could be indicative of the relative influence of selection vs. demographic processes in shaping genetic differentiation and population structure (Chen et al. 2012; Li et al. 2012). Genes showing allele frequencies that correlate with environmental clines are considered candidates for selection (outlier loci) (Hohenlohe et al. 2010; Pardo-Diaz et al. 2015). These footprints in the genome can be attributed to adaptation, but either the ascertainment of their association with the environmental variable under evaluation or its direct link with increasing fitness (phenotype) represents a challenge to be unravel. The main goal of this study was to analyse trends in genetic diversity in Nothofagus populations along precipitation gradients, using a suitable combination of markers. To accomplish this objective, variation at genes involved in response to drought tolerance, as well as microsatellites isolated from the transcriptome and located near functional genes (EST-SSRs), was examined in order to look for departures from neutrality. In addition, a set of microsatellites isolated from non-coding genomic regions were used to decouple the incidence of demographic events in the genetic structure. For this purpose, we selected two longitudinal transects along which precipitation decreases from west to east. In the northern transect (Lácar Lake, 40° S), the closely related N. nervosa and N. obliqua grow in sympatry. The southern transect (at 43° S) contains the more xeric forest patches of N. pumilio towards the east. Our testing hypothesis is that the establishment of a precipitation gradient shapes population genetic variation, although its major configuration was settled during last glaciations. We expect that populations located at the extremes (western or eastern edges) would be genetically different at candidate genes related to drought stress response; also that an unusual variation pattern of some genes that departs from neutral expectations could evidence ongoing adaptation processes to local conditions, more humid to the west and more xeric to the east. Between species, deeper genetic differentiation may be found, either reflecting macro-evolutionary changes due to the speciation process or their marked ecological preferences. The inclusion of three native species belonging to a key genus of the southern hemisphere may give clues for prediction of differential adaptive responses.
2 Material and methods
2.1 Environmental gradients and species under study
The three Nothofagus species under study are wind-pollinated, outcrossing, deciduous trees that hybridize naturally. Whereas N. pumilio is widely distributed in Patagonia (from 36 to 55° S), N. nervosa and N. obliqua are restricted to 36–40° S and are mostly protected within National Parks (Sabatier et al. 2011). The three species co-occur at some locations, but have different ecological niches towards the eastern edge. Therefore, we chose the two wider geographical ranges (at 40° S and 43° S) covering a decreasing precipitation regime. The main life-threatening period is the dry season (December to February, coincident with late spring and early summer seasons); in the annual cycle, this mostly coincides with the growing period of the species. Consequently, trees could be affected by drought stress, producing a differential effect along the west-east gradient.
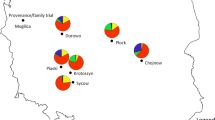
Plant material was collected from seven and six populations of N. nervosa and N. obliqua, respectively, totalling 251 and 208 individuals (average: 35 ind/pop) in the Lácar Lake watershed (40° S). Sampling covered from humid sites near the Andes (western populations, up to 2500 mm/year) to mesic sites towards the eastern shores of the lake (1200 mm/year, Fig. 1a, b), within a short distance (approximately 28 km; Table 1). A set of six N. pumilio populations were sampled at southern latitudes (Chubut region, 43°), with 236 trees (average: 39 ind/pop) (Table 1; Fig. 1c), along a rainfall gradient varying from humid sites (1800 mm/year) to the xeric edge of the distribution (200 mm/year). Populations P1 and P5 represent the extremes of this gradient, respectively. Eastern forests are fragmented, occurring in a steppe-like landscape with severe conditions throughout the year. A minimum distance of 30–50 m was maintained between individual sampled trees to avoid collecting full or half siblings.

Sampled locations of N. nervosa and N. obliqua in the Lácar Lake watershed (a, b, respectively) and of N. pumilio in the Vintter Lake watershed (c). Variation at microsatellites is represented on the maps by pie charts of proportional size according to heterozygosis estimations, both at gSSRs and EST-SSRs
2.2 DNA extraction and microsatellite amplification
Total DNA was extracted from dormant buds following Dumolin et al. (1995) with minor modifications, such as the replacement of mercaptoethanol with DTT 2%.
Nuclear microsatellites isolated from the non-coding genome (16, from now on named as genomic or gSSRs) and the transcriptome (11, from now on named as EST-SSRs) were amplified using either labelled primers (fluorescent mark incorporated in forward or reverse primer) or M13 methodology (Schuelke 2000) (Table S1, S2). Primer sequences, polymerase chain reaction (PCR) conditions and thermal profiles have been previously reported (gSSRs: Azpilicueta et al. 2004, Marchelli et al. 2008, Soliani et al. 2010, El Mujtar et al. 2014; EST-SSRs: Torales et al. 2012, El Mujtar et al. 2017). Individual genotypes were read on ABIPRISM 3700 capillary sequencer and allele assignment was carried out with GeneMapper v.3.7 (Applied Biosystem).
2.3 Candidate gene selection and sequencing
Candidate genes related to water use efficiency or drought stress tolerance were identified based on the functional annotation of N. nervosa transcriptome (Torales et al. 2012), performed from de novo assemblies. Candidates were selected using key terms from Gene Ontology (GO) (like ‘drought stress’, ‘water deprivation’, ‘response to stress’, ‘response to water’, ‘response to osmotic stress’, ‘water channel activity’, ‘water transport’), and based on reported gene families already studied in model or other tree species (e.g. chloroplastic drought-induced stress protein, dehydrin/ABA response, ethylene signalling protein, 4-coumarate-CoA ligase activity). Sequences of the selected genes (25) (Table S3) from different species, including N. nervosa, were recovered from The National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/(NCBI) database, or the Fagaceae Genomics Web, https://www.hardwoodgenomics.org/ (e.g. Fagus sylvatica, Quercus spp.), to create multiple alignments as references. Primers were designed around conserved regions in the target N. nervosa isotigs; this strategy allowed consideration of inter-specific homology of genes. We used Primer3 (Untergasser et al. 2007) and then OligoAnalyzer 3.1 (http://www.idtdna.com/calc/analyzer) to check primer properties (e.g. melting temperature, formation of dimers). Oligonucleotides were validated on genomic DNA, following a general touchdown PCR procedure with modifications needed for each fragment (mainly annealing temperature (Ta) and MgCl2 concentration). Post-processing of sequences before data analysis and detailed information of inspected genes are reported in the supporting online information (see Table S3).
2.4 Data analysis
2.4.1 Characterization of population genetic diversity and structure by microsatellites
To compare differentiation due to demographic or selection processes, we first used microsatellites. Genetic diversity and structure was inferred from microsatellite markers considering (i) genomic SSRs (totalling 16 gSSRs), presumed as selectively neutral, and (ii) transcriptome SSRs (totalling 11 EST-SSRs), supposed as putatively adaptive due to their proximal location to coding genes. Functional annotations of genes located in the near vicinity of EST-SSRs are detailed in Table S2. Seven N. nervosa populations and six N. obliqua populations (Fig. 1; Table S1) were genotyped using 14 SSRs (10 gSSR and 4 EST-SSR (Table S1). In N. pumilio from the seven gSSRs (Soliani et al. 2015) genotyped at six populations, one is common with the other species (Npum9) while from the eight EST-SSRs (Table S1), genotyped in three populations, one is common with the other two species (19SN).
Intra-species genetic diversity and differentiation were estimated by calculating allelic richness (AR) after rarefaction to a common sample size (El Mousadik and Petit 1996) in FSTAT (Goudet 2001), and the standardized differentiation index G’ST (Hedrick 2005) in GenAlEx (Peakall and Smouse 2006). Significant differences in genetic diversity (Nei’s unbiased heterozygosity and effective number of alleles) were checked through a test of multiple comparisons among populations with InfoGen (Balzarini and Di Rienzo 2004). Pairwise differentiation coefficients between populations and their significance after 1000 permutations were also obtained. An isolation-by-distance pattern was tested by comparing pairwise geographic (logarithm transformation) and Nei’s unbiased genetic distances between populations, using a Mantel test with 10,000 permutations to test for significance. A hierarchical analysis of molecular variance (AMOVA) was performed to evaluate the degree of genetic variation explained by (a) species (only between N. nervosa and N. obliqua that were genotyped with the same microsatellites), and (b) populations within species (GenAlEx 6.4.; Peakall and Smouse 2006). Statistical significance was obtained on the basis of 1000 permutations.
Genetic and geographic structure and admixture within species was assessed in STRUCTURE (Pritchard et al. 2000), based on the LOCPRIOR model (Hubisz et al. 2009), with admixture and correlated allele frequencies as described by Falush et al. (2003). Five independent runs for each K (from 1 to 10) were performed with a 10,000 burn-in period and 100,000 repetitions. The optimal number of clusters was obtained by the mean value of the log-likelihoods of 5 runs at each K (L K mean ± SD vs. K graph) and the rate of change in the log probability of data between successive K values (ΔK) (Evanno et al. 2005) using STRUCTURE HARVESTER (Earl and VonHoldt 2011). As expected, genomic SSRs that are usually more polymorphic than transcriptome SSRs show clearer genetic patterns between populations. The stability of genetic structure was tested by increasing the number of Ks (with respect to the optimum) on each species and marker type.
We additionally used discriminant analysis of principal components (DAPC) (Adegenet package in R) to find principal components that best fit two conditions: to summarize and detect differences between clusters and to minimize differences within clusters (Jombart et al. 2010). To identify the optimal number of clusters in this method, increasing values of K are run sequentially, and then the best grouping corresponds to the one with the lowest BIC. A correspondence between methods (STRUCTURE and DAPC) was found based on comparative clustering obtained with population’s membership. In agreement with the robustness of the results, a graphical representation was constructed based on both outputs. At the optimum clustering, we identified genetically homogeneous gene pools (i.e. shared clusters and similar membership coefficients at STRUCTURE, non-significant pairwise FST value), thus assumed to be in equilibrium for evolutionary forces altering demography. Accordingly, we selected individual trees from populations grouped in a homogeneous pool to sequence candidate genes (referred in Section 2.3) and evaluated its pattern of variation. The selected populations come from both edges of the respective precipitation gradients (Table 1; next section) in each species.
Additionally, we correlated mean annual precipitation (MAP) values with populations’ Nei’s unbiased heterozygosity by adjusting linear regressions. MAP was assumed as a proper proxy describing the established climate gradients since wet and dry seasons coincides in terms of duration and temporality at all sites, and therefore mean precipitation varies similarly along all gradients. Finally, we perform linear regressions between the most frequent alleles (> 10%) on each locus (at both gSSRs and EST-SSRs) and mean annual precipitation. These analyses and the p value for each adjusted r-squared (R2) were run on R environment (R Development Core Team 2011).
2.4.2 Characterization of candidate gene sequences related to drought stress
We analysed the populations at both edges of the respective gradients, two of N. nervosa, two of N. obliqua and two of N. pumilio, by re-sequencing from 6 to 12 polymorphic genes in 63 individual trees (totalling 126 haploid sequences). We screened up to 22 individuals from N. nervosa N7 and N2 populations (11 per population), 24 from N. obliqua O6 and O2 populations (12 per population) and 16 from N. pumilio P5 and P1 populations (8 per population). Nucleotide and haplotype diversity (π, θW, Hd) (Watterson 1975; Nei 1987), as well as number of segregating sites (S) and haplotypes (H) at population and species levels, were calculated for each gene in DNAsp v5 (Librado and Rozas 2009). To test for departures from the neutral model of evolution within populations, we ran different analytical methods: D (Tajima 1989), Fs (Fu 1997), R2 (Ramos-Onsins and Rozas 2002), D* and F* (Fu and Li 1993). Based on the coalescent algorithm, we ran computer simulations to calculate confidence intervals for each parameter, with segregating sites and no recombination with DNAsp v5 (Librado and Rozas 2009). We used the coalescent model because it is sensitive at detecting molecular changes in DNA when population expansion is occurring since it can be confounded with genetic hitchhiking giving similar genetic footprints (Ramos-Onsins and Rozas 2002; Librado and Rozas 2009). Mismatch distribution of expected vs. observed differences in segregating sites were checked for a population expansion model (population growth), and the raggedness statistic rg (Harpending 1994) was calculated and tested for significance. As evidences of population expansion, we expected a mismatch distribution plot as unimodal and negative (significant) values of D and Fs parameters. As indication of selection, we expected a multimodal distribution (stable population size) and positive D and R2 (neutrality test). We also estimated the number of synonymous substitutions per synonymous site (Ks) and divergence at non-synonymous substitutions sites (Ka) for any pair of sequences between species (for N. nervosa and N. obliqua), in coding regions, reporting an average value across genes.
Finally, single-nucleotide polymorphisms (SNPs) in all the sequenced gene regions were identified for each species and population. Linkage disequilibrium (LD) was evaluated in Genepop (Rousset 2008), and non-linked SNPs were used to test deviations from neutrality based on FST fixation index, as a multi-locus scan. Tests were based on the island model, assuming a common migration pool (Beaumont and Nichols 1996; Foll and Gaggiotti 2008), using Bayescan (Foll and Gaggiotti 2008) and LOSITAN (Antao et al. 2008). Bayescan is reliable for the detection of outliers from the distribution of locus-specific FST, with a low percentage of type I error (Narum and Hess 2011), and is suitable for small sample sizes. Departure from neutrality is assumed when the locus specific component of the inferred FST (alpha value) is significantly different from zero. Positive values of α suggest diversifying selection, whereas negative values suggest balancing or purifying selection. After running the MCMC algorithm with the default parameters, the posterior probability of each marker was calculated to infer the probability of selection over the neutral model. For the detection of outliers, we set a threshold to control for the false discovery rate (FDR), of 5%. In turn, LOSITAN evaluates the relationship between FST and HE, describing their expected distributions under an island model of migration with neutral markers. The distribution is then used to identify outlier candidates (higher or lower FST values compared to neutral expectations) for selection (Antao et al. 2008). LOSITAN was also used to test for departures from mutation-drift equilibrium at multi-locus genotypes at microsatellites in all species. In N. pumilio populations, this was of particular interest because contrasting genetic trends of ESTs with respect to gSSRs might indicate the action of different evolutionary forces shaping variation in this species.
3 Results
3.1 Genetic diversity and differentiation at microsatellites
Genomic markers (gSSRs) were more polymorphic than transcriptome microsatellites (EST-SSRs) in all three species, having more alleles and higher levels of allelic richness (Table 1). Differentiation coefficients, both across populations FST and G’ST (Table 1), and pairwise coefficients (FST) (Table S4) were higher for gSSRs than for EST-SSRs in all species. However, the hierarchical AMOVA showed that the proportion of genetic variance partitioned between the species (N. nervosa, N. obliqua) was 34% for gSSRs (FRT = 0.34, p = 0.001) and 46% for EST-SSRs (FRT = 0.46, p = 0.001, Table S4). On the other hand, in each species, the greater percentage of variance was explained by variation within populations (91–98%), and to a lesser extent by variation among populations (FST ~ 2–9%) (Table S4).
N. nervosa and N. obliqua showed different genetic diversity trends at both genomic and transcriptome markers in Lácar Lake (Table 1, Fig. 1). N. nervosa showed higher variation to the west at both set of markers (at EST-SSRs HE for N5 significantly higher than HE for N1-N4 populations, and increasing values from east to west in gSSRs, although non-significant). In the case of N. obliqua, the tendency was less clear, but the eastern populations were more variable showing a decreasing diversity from O1 to O5 that raised up again in O6 for both marker sets (Table 1, Fig. 1). In N. pumilio, opposing trends were found for the markers: higher genetic diversity for gSSRs at western populations (P2-P6) and to the east for EST-SSRs (P1) (Fig. 1).
Population genetic diversity significantly correlated with mean annual precipitation in N. nervosa for EST-SSRs markers and a linear regression between variables was adjusted (R2 = 0.51, p = 0.044) (Fig. 2a). Moreover, allele frequencies and precipitation were found to be significantly related for alleles of NgBio111 (gSSR), and for 19SN, 13SN and 8SN (EST-SSR loci) in N. nervosa (p < 0.05), and for Npum13 (gSSR; p < 0.05) in N. pumilio (Fig. 2b) but this was not found in N. obliqua. By doing a locus specific analysis of molecular variance on each species, we detected some SSR loci with particularly high values of differentiation (> 10%) among populations (Notho214 in N. nervosa; NnBio37, Npum14, Notho214, 23SN in N. obliqua; Npum13 in N. pumilio). The Mantel test was not significant for any of the three species or markers and the isolation-by-distance pattern was not supported.

Geographic genetic variation expressed by the correlations between genetic diversity (HE) or allele frequencies and mean annual precipitation (MAP) along precipitation gradients. Graphs were designed by species and loci, and only significant correlated relationships (p ≤ 0.05) are shown. a) Correlation between average values of population genetic diversity (HE; EST-SSRs) and population’s mean annual precipitation (MAP) values in N. nervosa; b) Correlation between allele frequencies by population (gSSR or EST-SSRs) vs. population’s mean annual precipitation values in N. nervosa and N. pumilio
3.2 Population structure
In all three species, discriminant analysis (DAPC) and STRUCTURE bring about a similar grouping for both gSSRs and EST-SSRs, when pre-defined populations were set (Fig. 3). Divergent populations (i.e. singular cluster) within each species were recognized. In addition, clustering methods revealed the admixed nature of several populations with variable percentage of membership assigned to different clusters. It should be stressed that both methods provide the same clustering. Optimum K and two consecutive clustering plots from STRUCTURE were represented for each species and marker: in all species, gSSRs showed optimum clustering at K = 3, whereas in EST-SSRs K = 3 in N. nervosa and N. pumilio and K = 2 in N. obliqua were the optimum grouping (Fig. 3, Fig. S1).

Genetic structure in the evaluated populations represented as individual membership coefficients for genetic demes with Bayesian clustering (STRUCTURE), and discriminant analysis of principal components (DAPC), for genomic SSRs (A-1, B-1, C-1) and EST-SSRs (A-2, B-2, C-2). The optimum and two consecutive Ks are shown for STRUCTURE clustering in each species and markers. For better visualization, populations are ordered from west to east in the Bayesian clustering output. A: N. nervosa; B: N. obliqua; C: N. pumilio. Population labels coincide with Table 1
3.3 Nucleotide diversity and neutrality tests based on coalescent simulations
We found 50 and 43 SNPs within N. nervosa and N. obliqua populations, respectively, in 12 polymorphic candidate genes involved in water stress resistance (average length 551-559 bp, Table S3), while between species we found 146 SNPs. In N. pumilio, we detected 42 SNPs in seven polymorphic genes (average length 534 bp, Table S3).
At species level, nucleotide diversity was higher in N. obliqua than in N. nervosa, with a total average value of πt = 0.00226 vs. 0.00155 and Hd = 0.4838 vs. 0.402 (Table 2). A higher diversity value was found for the eastern N. nervosa and for the western N. obliqua populations, respectively (Table 2). Divergence at synonymous sites (Ks) was greater in the majority of genes than divergence at non-synonymous sites (Ka) (mean values Ks = 0.02139, Ka = 0.0072) with some exceptions (Ks < Ka in AdHy, Lea2 and PP2C-3). In N. pumilio, nucleotide diversity showed an average value of πt = 0.0028 and Hd = 0.619. A higher diversity value was found in the western population of the species (Table 2). Nei’s (πt) and Watterson’s (θwt) nucleotide diversity values per gene and population are presented in Table S5.
From the 13 candidate genes analysed (6 of which were common to all species, Table S3, S5), in 6 populations of 3 species we reported in detail the cases in which significant tests allow us to inferred signals of demography and/or selection. We detected several genes showing the signal of demographic changes (unimodal mismatch distribution, not significant raggedness statistic, negative D and Fs values; Table 3 Fig. S2). We observed this pattern displayed by three genes (ß-amylase protein (BAM1), ethylene signalling protein (Ethyl) and late embryogenesis abundant protein (Lea2)) in the N. nervosa western population (N7) and four genes (BAM1, Lea2 and two protein phosphatase (PP2C1 and PP2C3)) in N. obliqua eastern population (O2), which could be interpreted as an evidence of population expansion. In turn, one gene (chromatin-remodelling (ATCHR12)) exhibited the same trend in O6 (N. obliqua) population. Meanwhile, in N. pumilio eastern population, we found a similar situation in two genes (Ethyl and protein kinase (SOS2)). In sum, the effect of natural selection was not discernible although the genes presented significant values of R2 at neutrality tests (Table 3).
On the other hand, we found evidence of balancing selection in two populations of N. obliqua (O2 and O6) for chloroplastic drought-induced stress protein (Cdsp32) gene. Only in this case, neutrality test by means of D showed positive and significant results (Table 3) and the multimodal mismatch distribution of pairwise differences at sequence level in both populations supported the finding (Fig. S2).
3.4 Neutrality tests based on FST fixation index
From the multiscan approach, the average FST value across SNPs obtained with Bayescan was 0.083 for N. nervosa (varying from 0.080 (PP2C1) to 0.087 (Lea2)), 0.052 in N. obliqua (varying from 0.051 (SOS2) to 0.059 (PP2C3)) and 0.153 in N. pumilio (varying from 0.148 (heat shock protein (Hsp90) to 0.167 (PP2C1)).
LOSITAN detected several SNPs outside the expected neutral envelope. Some had higher FST values (candidates for positive selection) and others had lower FST values than expected (candidates for balancing selection) in each species (Fig. 4). Bayescan did not identify outliers in any of the species (p < 0.05, FDR test) (Fig. S3).

Analysis of FST outliers using LOSITAN a in N. nervosa, bN. obliqua, and cN. pumilio. Estimates of FST for each candidate were plotted against heterozygosity. FDR was set at 0.05. Higher than expected FST values (candidates for positive selection): Lea21, PP2C111, PP2C323, PP2C327, PP2C328 and RCD13, RCD14, RCD19 for N. nervosa; PP2C33 ATCHR128 and ATCHR1212 for N. obliqua; SOS23, Lea27, PP2C12 in N. pumilio. Lower than expected FST values (candidates for balancing selection): Cdsp322, Ethyl3, Ethyl6, Lea23, Lea24, PP2C13, PP2C15, PP2C37, PP2C325, AdHy1, BAM19 and RCD111 for N. nervosa; SOS25, SOS210, Ethyl8, Lea28, Lea211, PP2C319, PP2C322, PP2C325, PP2C326, PP2C328, AdHy3, AdHy5 and PhII1 for N. obliqua; Cdsp321, SOS25, SOS26, Ethyl1, Lea25, PP2C11, PP2C14, Hsp903, Hsp904 in N. pumilio
In addition, departure from neutral expectation was found in some EST-SSRs and gSSRs with LOSITAN. In N. pumilio, the significant values of the differentiation coefficient in IN1207a and Npum9 were interpreted as signs of balancing selection, whereas IN0192a, Npum10 and Npum18 with values significantly above zero were considered as targets of positive selection (Fig. S4). In N. nervosa and N. obliqua, two gSSR markers that departed from neutrality were common to both species (Notho214, NnBio37). Additionally, departures were found in other markers either gSSRs (NnBIO11 N. nervosa; Npum14 N. obliqua) or EST-SSRs (23SN N. nervosa; 19SN N. obliqua). Not only the action of selection but also the presence of null alleles (checked in Azpilicueta et al. 2013, Soliani et al. 2015) could be determining this result (Fig. S4).
4 Discussion
4.1 Population demographic history across the precipitation gradient
Current genetic variation of the three Nothofagus species studied here still shows the footprints from Pleistocene glaciations (Marchelli et al. 1998; Marchelli and Gallo 2006; Azpilicueta et al. 2009; Soliani et al. 2015), reinforcing the hypothesis that these events were a crucial driving factor of genetic diversity. Our results agree with trends of locally common alleles (LCA) and with the distribution of cpDNA haplotypes variation (Marchelli et al. 1998; Marchelli and Gallo 2006; Azpilicueta et al. 2009), where higher diversity was detected to the west for N. nervosa and to the east for N. obliqua. All these results combined provide evidences of cryptic refugia locations: westward for N. nervosa (higher diversity in gSSRs and EST-SSRs) and N. pumilio (higher diversity in gSSRs), and eastward for N. obliqua (higher diversity in gSSRs and EST-SSRs).
During the last glaciations, there were periods of isolation in which the contraction and expansion of the populations might have occurred, and this might have shaped the genetic stocks of the species as has been suggested for several other plant and animal species from Patagonia (Sérsic et al. 2011; Turchetto-Zolet et al. 2013). Both demographic expansion within refugia and expansion from refugia towards colonized sites (i.e. range expansions) might have occurred. Still the presence of Pleistocene alpine valley glaciers that created present-day watersheds (Markgraf et al. 1996; Glasser et al. 2008), greatly determined the chance of demographic growth. When ice lobes retracted, migration pathways opened and re-colonization patterns from multiple refugia were settled (e.g. Marchelli et al. 1998) and therefore many populations within each species have a common origin and share genetic clusters (Fig. 3). The admixed populations that we found in this study gives support to the confluence of migration routes along the watersheds. We suggest that the range expansion from hotspots of diversity and the admixture due to meeting points after colonization (from projected species distribution during the last glacial maximum (LGM), Marchelli et al. 2017) are the reasons that gave rise to the observed genetic gradient. Based-evidence of the latter is the N. pumilio population P3, which is located at the centre of an admixed zone (Soliani et al. 2015), has the highest allelic richness value and two genetic clusters. In addition, the easternmost population (P1), which is a marginal population with patches of individuals distributed across a steppe-like environment, presented a higher proportion of a particular cluster at successive Ks for gSSRs in relation to the other populations (Fig. 3). This population also showed low genetic diversity values. Its isolation during past glaciations is evidenced by a different genetic composition and greater differentiation due to evolutionary forces (e.g. genetic drift, selection pressure or the effects of a genetic bottleneck).
On the other hand, the unveiled correlation between genetic variation and precipitation might provide clues about processes driving more recent population evolution. Specifically, the significant relationship between MAP and allele frequencies (both coming from gSSRs and EST-SSRs, Fig. 2b) and Nei’s unbiased heterozygosis (from EST-SSRs, Fig. 2a), nourish the existence of clinal variation in N. nervosa and N. pumilio. In addition, opposite tendencies in allele frequencies along climate gradients are harboured at loci with particularly high differentiation coefficients. These patterns were interpreted in two alternate ways. First, if alleles at neutral loci tend towards fixation at the extreme edges, it could be evidence of colonization (e.g. due to founder effect). If two different genetic lineages came into secondary contact (e.g. Durand et al. 2009), a similar trend in allele frequencies might also be established after admixture. Postglacial migration may have promoted clinal variation, determining spatial patterns along the current gradients, further confounding putative selection effects (Kujala et al. 2017). Alternatively, clines may be interpreted as being consistent with adaptation, which is particularly hard to unravel in the case of alleles coming from transcriptome SSRs (ESTs) (Table S2). In these markers, there is a risk of interpreting the random effects of population dynamics with the selection of adapted variants (Tiffin and Ross-Ibarra 2014; Roschanski et al. 2016) due to the hitchhiking effect (Stinchcombe and Hoekstra 2007). Signs of demographic changes might confound the action of selection on the inspected candidate genes; therefore, an unequivocal interpretation of adaptation at local level should be dismissed. In order to decouple demography from the role of selection in generating adaptive variants along gradients, we will further discuss candidate genes’ variation trends.
4.2 Trends of genetic diversity at candidate genome regions along the rainfall gradient
Our work show evidences supporting that selection pressure caused by varying precipitation regimes could be one of the driving factors promoting different trends at candidate genes. We detected significant signals of balancing selection in Cdsp32 gene in two N. obliqua populations. Although this signal was not significant in N. nervosa and N. pumilio, this gene did present higher nucleotide diversity in all species in the eastern populations of their respective ranges, i.e. more xeric locations (Table S5). Even acknowledging the need of more populations finally confirm this pattern, the higher intra-population genetic diversity detected might favour populations’ evolution by natural selection. Thus, we presume that the selective pressures imposed in these gradually changing environments may involve a relevant contribution shaping the previously established patterns of genetic diversity (e.g. Azpilicueta et al. 2009; Marchelli et al. 2017; Soliani et al. 2015), in agreement with our hypothesis. In such highly selective environments, which have conditions comparable to those predicted under global climate change scenarios (Rusticucci and Barrucand 2004; Castañeda and González 2008), adaptation might be improved either by the fixation of local variants or the immigration of more suitably adapted alleles (i.e. regional gene flow).
However, we were not able to distinguish signals of selection in other genes. Although neutrality tests (R2) in both BAM1, Lea2 in N. nervosa and N. obliqua, or Ethyl in all species were significant, the variation could not be unequivocally attributed to current selection pressures (unimodal distribution, non-significant raggedness statistic, Fig. S2). The result suggests a non-equilibrium condition that could be attributed to demographic population expansion after establishment (i.e. post-glaciations). The legacy of ancient marks in genetic variation is probably inherited from an ancestral refugial population, which also supports our hypothesis.
The functional aspects of stress resistance genes (Cdsp32, PhII and BAM1) are worth mentioning because of their role involved in water use in chloroplast metabolism. By assuming that functional mechanisms are conservative in the plant kingdom, we can use homologue fragments of our candidate genes from Arabidopsis and from phylogenetically related tree species (e.g. Quercus or Fagus) as a reference to Nothofagus. The chloroplastic drought-induced stress protein (Cdsp32) is known to be regulated by environmental and oxidative stress conditions (Broin et al. 2002). Encoded in the nucleus and located in the stroma, its production is abundant when plants are under stress (Broin et al. 2002; Wang et al. 2014). In Arabidopsis, the Chlorophyll a/b binding protein-photosystem II light harvesting complex (PhII) plays a positive role in abscisic acid signalling, in part by modulating reactive oxygen species (ROS) homeostasis (Xu et al. 2012). BAM1 degrades transitory starch to sustain proline biosynthesis during drought stress (Zanella et al. 2016), suggesting that BAM1 plays a role in water use efficiency.
Even if not conclusive about an adaptive response, some EST-SSRs showed a departure from the neutral envelope. In N. pumilio, functional annotation of the IN01207a and IN00192a transcriptomic SSRs matches coding regions of heat shock proteins (N. nervosa transcriptome, Torales et al. 2012) and ‘response to stimulus’ Gene Onthology term. Since the natural range of the cold-tolerant species N. pumilio is restricted to high latitudinal and elevation ranges, our result suggests potential sensitivity of the species to rising temperatures and desiccation. For example, the marginal population P1, which is located in an environment with higher mean temperatures and less precipitation, presented higher diversity at the IN01207a locus. In N. nervosa and N. obliqua, 23SN and 19SN markers were found near genes in response to osmotic stress and metal ion/DNA repair, respectively. Ionic toxicity within cells due to salt accumulation could be indirect evidence of drought. In the long term, salinity could produce oxidative stress in plants with the generation of reactive oxygen species (ROS) (e.g. You and Chan 2015). Further inspection of all these gene regions could construct a more complete picture of adaptive variation in the species of interest.
Species trends in genetic variation support distinctive evolutionary histories as well as an own adaptive potential, which is signalled by the different ecological preferences. The higher genetic diversity in candidate genes observed in N. obliqua might be related to its ability to occupy a wider geographical range along precipitation gradients in Patagonia (longitudinal range of N. nervosa and N. obliqua are 71.40° W–71.19° W and 71.40° W–70.57° W, respectively). Over time, different adaptive variants might lead to fixation within each species following their physiological optima (i.e. water use efficiency, Varela et al. 2012), and potentially may even increase genetic divergence. Differential responses along their environmental distribution ranges may explain a genetic control that shapes species’ expressed traits (e.g. Bourne et al. 2017). A greater number of genes and populations within species would allow us to delve deeper into intraspecific divergence, to better elucidate how adaptation occurs.
Strategies for adapting to environmental changes include local adaptation, plasticity, or distributional shifts along climate gradients (e.g. Aitken et al. 2008). Moreover, colonizing new and more suitable habitats might reduce extinction risk. Particularly in Nothofagus species, which have limited seed (e.g. Rusch 1987) and pollen dispersal (e.g. Marchelli et al. 2012) and low seedling recruitment and sapling survival (e.g. Cuevas 2002), populations’ resilience and recovery might be compromised. Although these factors may reduce the likelihood of succeed, local adaptation is still possible. Considering species’ ecological characteristics, future suitability predictions suggest a better outlook for N. obliqua than for N. nervosa, reflecting different tolerance levels to the severe climatic conditions expected (Marchelli et al. 2017). Additionally, ecological niche modelling of N. pumilio predicts the retraction of forests in the westernmost portions of its current distribution range (Soliani et al. in prep.) threatening border populations.
5 Conclusions
Species’ genetic trends at neutral markers and signs of population expansion in functional regions (unimodal mismatch distribution, negative D and Fs coefficients), probably associated to re-colonization, highlights the fact that footprints from the recent past are still deeply imprinted in Nothofagus gene pools. The correlation of genetic diversity with precipitation, and signs of balancing selection in outlier candidates, constituted preliminary evidence of gene regions functionally related to key responses of trees inhabiting environmental gradients. Candidate genes evaluated here could be part of the multiple loci that mediate adaptation to drought stress and modulate plant fitness in these species. We have taken a first step towards understanding how selection acts on the precipitation distribution range of South American Nothofagus. This knowledge could provide a base for inferring future species behaviour in the context of adaptation to changing environmental conditions. In addition, this information is essential to delineate current conservation, mitigation or management plans.
Data availability
Individual gene sequences were deposited to GeneBank (https://www.ncbi.nlm.nih.gov/). Accession numbers MF446356-MF446390; Multilocus SSRs genotypes, candidate genes genotypes and the supplementary material were deposited in Zenodo repository (Soliani et al. 2019): https://doi.org/10.5281/zenodo.3544568
References
Aitken SN, Yeaman S, Holliday JA, Wang T, Curtis-McLane S (2008) Adaptation, migration or extirpation: climate change outcomes for tree populations. Evol Appl 1:95–111. https://doi.org/10.1111/j.1752-4571.2007.00013.x
Alberto FJ, Derory J, Boury C, Frigerio JM, Zimmermann NE, Kremer A (2013) Imprints of natural selection along environmental gradients in phenology-related genes of Quercus petraea. Genetics 195:495–512. https://doi.org/10.1534/genetics.113.153783
Alexander LV, Zhang X, Peterson TC et al (2006) Global observed changes in daily climate extremes of temperature and precipitation. J Geophys Res 111:D05109. https://doi.org/10.1029/2005jd006290
Allen CD, Macalady AK, Chenchouni H et al (2010) A global overview of drought and heat-induced tree mortality reveals emerging climate change risks for forests. For Ecol Manag 259:660–684. https://doi.org/10.1016/j.foreco.2009.09.001
Antao T, Lopes A, Lopes R et al (2008) LOSITAN: a workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9:323
Azpilicueta MM, Caron H, Bodénès C, Gallo LA (2004) SSR markers for analysing South American Nothofagus species. Silvae Genet 53:240–243
Azpilicueta MM, Marchelli P, Gallo LA (2009) The effects of quaternary glaciations in Patagonia as evidenced by chloroplast DNA phylogeography of southern beech Nothofagus obliqua. Tree Genet Genomes 5:561–571
Azpilicueta MM, Gallo LA, van Zonneveld M et al (2013) Management of Nothofagus genetic resources: definition of genetic zones based on a combination of nuclear and chloroplast marker data. For Ecol Manag 302:414–424. https://doi.org/10.1016/j.foreco.2013.03.037
Balzarini M, Di Rienzo J (2004) Info-Gen: Software para análisis estadístico de datos genéticos. Facultad de Ciencia Agropecuarias. Universidad Nacional de Córdoba, Argentina
Beaumont MA, Nichols RA (1996) Evaluating loci for use in the genetic analysis of population structure. Proc R Soc Lond Ser B Biol Sci 263:1619–1626. https://doi.org/10.1098/rspb.1996.0237
Bourne AE, Creek D, Peters JMR, Ellsworth DS, Choat B (2017) Species climate range influences hydraulic and stomatal traits in Eucalyptus species. Ann Bot 120:123–133
Broin M, Cuiné S, Eymery F, Rey P (2002) The plastidic 2-cysteine peroxiredoxin is a target for a thioredoxin involved in the protection of the photosynthetic apparatus against oxidative damage. Plant Cell 14:1417–1432. https://doi.org/10.1105/tpc.001644
Castañeda M, González M (2008) Statistical analysis of the precipitation trends in the Patagonian region in southern South America. Atmósfera 21:303–317
Centro de Investigaciones del Mar y de la Atmósfera (2015) Tercera comunicación nacional de la República Argentina a la convencion marco de las Naciones Unidas sobre el cambio climático
Chen J, Källman T, Ma X, Gyllenstrand N, Zaina G, Morgante M, Bousquet J, Eckert A, Wegrzyn J, Neale D, Lagercrantz U, Lascoux M (2012) Disentangling the roles of history and local selection in shaping clinal variation of allele frequencies and gene expression in Norway spruce (Picea abies). Genetics 191:865–881
Cosacov A, Sérsic AN, Sosa V et al (2010) Molecular evidence of ice-age refugia in the Patagonia steppe and post-glacial colonisation of the Andes slopes: insights from the endemic species Calceolaria polyrhiza (Calceolariaceae). J Biogeogr 37:1463–1477
Cosacov A, Johnson LA, Paiaro V et al (2012) Precipitation rather than temperature influenced the phylogeography of the endemic shrub Anarthrophyllum desideratum in the Patagonian steppe. J Biogeogr 40:168–182. https://doi.org/10.1111/j.1365-2699.2012.02776.x
Cuevas JG (2002) Episodic regeneration at the Nothofagus pumilio alpine timberline in Tierra del Fuego, Chile. J Ecol 90:52–60
Dixon CJ (2010) OLFinder—a program which disentangles DNA sequences containing heterozygous indels. Mol Ecol Resour 10:335–340. https://doi.org/10.1111/j.1755-0998.2009.02749.x
Dumolin S, Demesure B, Petit RJ (1995) Inheritance of chloroplast and mitochondrial genomes in pedunculate oak investigated with an efficient PCR method. Theor Appl Genet 91:1253–1256
Durand E, Jay F, Gaggiotti OE, Francois O (2009) Spatial inference of admixture proportions and secondary contact zones. Mol Biol Evol 26:1963–1973
Earl DA, VonHoldt BM (2011) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
El Mujtar VA, Gallo LA, Tiange L, Garnier-Géré P (2014) Development of genomic resources for Nothofagus species using next-generation sequencing data. Mol Ecol Resour 14:1281–1295. https://doi.org/10.1111/1755-0998.12276
El Mujtar V, López M, Amalfi S et al (2017) Characterization and transferability of transcriptomic microsatellites markers for Nothofagus species. New Zeal J Bot. https://doi.org/10.1080/0028825X.2017.1340310
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Molecular Ecology 14(8):2611–2620
Fady B, Cottrell J, Ackzell L et al (2016) Forests and global change: what can genetics contribute to the major forest management and policy challenges of the twenty-first century? Reg Environ Chang 16:927–939. https://doi.org/10.1007/s10113-015-0843-9
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164:1567–1587
Foll M, Gaggiotti O (2008) A genome-scan method to identify selected loci appropriate for both dominant and codominant markers: a Bayesian perspective. Genetics 180:977–993
Fu Y-X (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Fu Y-X, Li W-H (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Glasser NF, Jansson KN, Harrison S, Kleman J (2008) The glacial geomorphology and Pleistocene history of South America between 38oS and 56oS. Quat Sci Rev 27:365–390
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from https://www2.unil.ch/popgen/softwares/fstat.htm. Accessed 16 Dec 2019
Grivet D, Sebastiani F, Alia R et al (2011) Molecular footprints of local adaptation in two Mediterranean conifers. Mol Biol Evol 28:101–116
Harpending H (1994) Signature of ancient population growth in alow-resolution mitochondrial DNA mismatch distribution. Hum Biol 66:591–600
Hedrick PW (2005) A standardized genetic differentiation measure. Evolution (N Y) 59:1633–1638
Heuertz M, De Paoli E, Källman T et al (2006) Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 174:2095–2105
Hohenlohe PA, Phillips PC, Cresko WA (2010) Using population genomics to detect selection in natural populations: key concepts and methodological considerations. Int J Plant Sci 171:1059–1071. https://doi.org/10.1086/656306
Hubisz MJ, Falush D, Stephens M, Pritchard JK (2009) Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources 9(5):1322–1332
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. https://doi.org/10.1186/1471-2156-11-94
Kawecki TJ, Ebert D (2004) Conceptual issues in local adaptation. Ecol Lett 7:1225–1241
Kremer A, Ronce O, Robledo-Arnuncio JJ, Guillaume F, Bohrer G, Nathan R, Bridle JR, Gomulkiewicz R, Klein EK, Ritland K, Kuparinen A, Gerber S, Schueler S (2012) Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol Lett 15:378–392
Kujala ST, Knurr T, Karkkainen K et al (2017) Genetic heterogeneity underlying variation in a locally adaptive clinal trait in Pinus sylvestris revealed by a Bayesian multipopulation analysis. Heredity (Edinb) 118:413–423
Li J, Li H, Jakobsson M, Li S, Sjödin P, Lascoux M (2012) Joint analysis of demography and selection in population genetics: where do we stand and where could we go? Mol Ecol 21:28–44. https://doi.org/10.1111/j.1365-294X.2011.05308.x
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452
Marchelli P, Gallo LA (2006) Multiple ice-age refugia in a southern beech from southern South America as revealed by chloroplast DNA markers. Conserv Genet 7:591–603
Marchelli P, Gallo L, Scholz F, Ziegenhagen B (1998) Chloroplast DNA markers reveal a geographical divide across Argentinean southern beech Nothofagus nervosa (Phil.) Dim. et Mil. distribution area. Theor Appl Genet 97:642–646
Marchelli P, Caron H, Azpilicueta MM, Gallo L (2008) Primer note: a new set of highly polymorphic nuclear microsatellite markers for Nothofagus nervosa and related South American species. Silvae Genet 57:82–85
Marchelli P, Smouse P, Gallo L (2012) Short-distance pollen dispersal for an outcrossed, wind-pollinated southern beech (Nothofagus nervosa (Phil.) dim. Et mil.). Tree Genet Genomes 8:1123–1134
Marchelli P, Thomas E, Azpilicueta MM, Zonneveld M, Gallo L (2017) Integrating genetics and suitability modelling to bolster climate change adaptation planning in Patagonian Nothofagus forests. Tree Genet Genomes 13:119–114. https://doi.org/10.1007/s11295-017-1201-5
Markgraf V, Romero EJ, Villagrán C (1996) History and paleoecology of South American Nothofagus forests. In: Veblen TT et al (eds) In: the ecology and biogeography of Nothofagus forests. Yale Univ Press, New Haven London, pp 354–386
Masiokas MH, Villalba R, Luckman BH et al (2008) 20th-century glacier recession and regional hydroclimatic changes in northwestern Patagonia. Glob Planet Chang 60:85–100
Mathiasen P, Premoli AC (2010) Out in the cold: genetic variation of Nothofagus pumilio (Nothofagaceae) provides evidence for latitudinally distinct evolutionary histories in austral South America. Mol Ecol 19:371–385
Mosca E, Eckert AJ, Liechty JD, Wegrzyn JL, la Porta N, Vendramin GG, Neale DB (2012) Contrasting patterns of nucleotide diversity for four conifers of Alpine European forests. Evol Appl 5:762–775. https://doi.org/10.1111/j.1752-4571.2012.00256.x
Narum S, Hess J (2011) Comparison of FST outlier tests for SNP loci under selection. Mol Ecol Resour 11:184–194. https://doi.org/10.1111/j.1755-0998.2011.02987.x
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Pardo-Diaz C, Salazar C, Jiggins CD (2015) Towards the identification of the loci of adaptive evolution. Methods Ecol Evol 6:445–464. https://doi.org/10.1111/2041-210X.12324
Peakall R, Smouse P (2006) GENALEX 6: genetic analysis in excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Pluess AR, Frank A, Heiri C et al (2016) Genome–environment association study suggests local adaptation to climate at the regional scale in Fagus sylvatica. New Phytol 210:589–601. https://doi.org/10.1111/nph.13809
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
R Development Core Team R (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna ISBN 3–900051–07-0, URL http://www.R-project.org/
Ramos-Onsins S, Rozas J (2002) Statistical properties of newneutrality tests against population growth. Mol Biol Evol 19:2092–2100
Roschanski AM, Csilléry K, Liepelt S, Oddou-Muratorio S, Ziegenhagen B, Huard F, Ullrich KK, Postolache D, Vendramin GG, Fady B (2016) Evidence of divergent selection for drought and cold tolerance at landscape and local scales in Abies alba Mill. in the French Mediterranean Alps. Mol Ecol 25:776–794. https://doi.org/10.1111/mec.13516
Rousset F (2008) GENEPOP’007: a complete re-implementation of GENEPOP software for Windows and Linux. Mol Ecol Notes 8:103–106
Rusch V (1987) Estudio sobre la regeneración de la lenga en la Cuenca del Río Manso Superior, Río Negro. Informe interno, CONICET, p 112
Rusticucci M, Barrucand M (2004) Observed trends and changes in temperature extremes over Argentina. Am Meteorol Soc 17:4099–4107
Sabatier Y, Azpilicueta MM, Marchelli P et al (2011) Distribución natural de Nothofagus alpina y Nothofagus obliqua (Nothofagaceae) en Argentina, dos especies de primera importancia forestal de los bosques templados norpatagónicos. Boletín la Soc Argentina Botánica 46:131–138
Savolainen O, Kujala ST, Sokol C et al (2011) Adaptive potential of northernmost tree populations to climate change, with emphasis on scots pine (Pinus sylvestris L.). J Hered 102:526. https://doi.org/10.1093/jhered/esr056
Schuelke M (2000) An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol 18:233–234
Sérsic A, Cosacov A, Cocucci AA et al (2011) Emerging phylogeographical patterns of plants and terrestrial vertebrates from Patagonia. Biol J Linn Soc 103:475–494
Soliani C, Sebastiani F, Marchelli P et al (2010) Development of novel genomic microsatellite markers in the southern beech Nothofagus pumilio (Poepp. Et Endl.) Krasser. In: permanent genetic resources added to molecular ecology resources database 1 October 2009–30 November 2009. Mol Ecol Resour 10:404–408
Soliani C, Tsuda Y, Bagnoli F et al (2015) Halfway encounters: meeting points of colonization routes among the southern beeches Nothofagus pumilio and N. antarctica. Mol Phylogenet Evol 85:197–207. https://doi.org/10.1016/j.ympev.2015.01.006
Soliani C, Azpilicueta MM, Arana MV, Marchelli P (2019) Clinal variation along precipitation gradients in Patagonian temperate forests: unravelling demographic and selection signatures in three Nothofagus spp.V2. Zenodo. [dataset]. https://doi.org/10.5281/zenodo.3544568
Stephens M, Donelly P (2003) A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162–1169
Stinchcombe JR, Hoekstra HE (2007) Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity (Edinb) 100:158–170
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tiffin P, Ross-Ibarra J (2014) Advances and limits of using population genetics to understand local adaptation. Trends Ecol Evol 29:673–680. https://doi.org/10.1016/j.tree.2014.10.004
Torales SL, Rivarola M, Pomponio MF, Fernández P, Acuña CV, Marchelli P, Gonzalez S, Azpilicueta MM, Hopp HE, Gallo LA, Paniego NB, Poltri SN (2012) Transcriptome survey of Patagonian southern beech Nothofagus nervosa (= N. Alpina): assembly, annotation and molecular marker discovery. BMC Genomics 13:291. https://doi.org/10.1186/1471-2164-13-291
Turchetto-Zolet AC, Pinheiro F, Salgueiro F, Palma-Silva C (2013) Phylogeographical patterns shed light on evolutionary process in South America. Mol Ecol 22:1193–1213. https://doi.org/10.1111/mec.12164
Untergasser A, Nijveen H, Rao X, Bisseling T, Geurts R, Leunissen JA (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res 35:W71–W74. https://doi.org/10.1093/nar/gkm306
Varela SA, Fernández ME, Gyenge J et al (2012) Physiological and morphological short-term responses to loght and temperature in two Nothofagus species of Patagonia, South America. Photosynthetica 50:557–569
Villalba R, Masiokas MH, Kitzberger T, Boninsegna JA (2005) Biogeographical consequences of recent climate changes in the southern Andes of Argentina. Glob Chang Mt Reg UM Uber al Springer, Netherlands, pp 157–166
Villalba R, Lara A, Masiokas MH et al (2012) Unusual southern hemisphere tree growth patterns induced by changes in the southern annular mode. Nat Geosci 5:793–798
Wang X, Vignjevic M, Jiang D, Jacobsen S, Wollenweber B (2014) Improved tolerance to drought stress after anthesis due to priming before anthesis in wheat (Triticum aestivum L.) var. Vinjett. J Exp Bot 65:6441–6456. https://doi.org/10.1093/jxb/eru362
Watterson G (1975) On the number of segregating sites in geneticalmodels without recombination. Theor Pop Biol 7:256–276
Xu Y-H, Liu R, Yan L, Liu ZQ, Jiang SC, Shen YY, Wang XF, Zhang DP (2012) Light-harvesting chlorophyll a/b-binding proteins are required for stomatal response to abscisic acid in Arabidopsis. J Exp Bot 63:1095–1106
You J, Chan Z (2015) ROS regulation during abiotic stress responses in crop plants. Front Plant Sci 6:1092. https://doi.org/10.3389/fpls.2015.01092
Zanella M, Borghi GL, Pirone C, Thalmann M, Pazmino D, Costa A, Santelia D, Trost P, Sparla F (2016) β-Amylase 1 (BAM1) degrades transitory starch to sustain proline biosynthesis during drought stress. J Exp Bot 67:1819–1826
Acknowledgements
We thank Dr. Georgina Sola for kindly providing the microsatellite genotypes of the Quilanlahue Population, Fernando Umaña and Victoria Lantschner for drawing up the maps of sampled populations, Alejandro Aparicio for statistical support in R, Katharina Budde for critical reading of the manuscript and Jill T. Sekely for English editing.
Funding
This work was funded by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT - FONCyT), Argentina [grant numbers PICT 2011 N° 2250 and PICT 2013 N°0603].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Handling Editor: Ricardo Alia
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contribution of the co-authors
Carolina Soliani, María Marta Azpilicueta, María Verónica Arana and Paula Marchelli conceived and designed the experiments; Carolina Soliani performed the experiments, analyzed the data and wrote the first version of the manuscript and all authors contributed to the writing and revision of the final manuscript.
Rights and permissions
About this article

Cite this article
Soliani, C., Azpilicueta, M.M., Arana, M.V. et al. Clinal variation along precipitation gradients in Patagonian temperate forests: unravelling demographic and selection signatures in three Nothofagus spp.. Annals of Forest Science 77, 4 (2020). https://doi.org/10.1007/s13595-019-0908-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13595-019-0908-x