
Abstract
Multiple ion transition summation of isotopologues (MITSI) is an adaptable and easy-to-implement methodology for improving analytical sensitivity, especially for halogenated compounds and otherwise abundant isotopologues. This novel application of signal summing was applied to measure and quantitate the two most abundant ion transitions of two isotopologues of N-acetyl-S-(1,2-dichlorovinyl)-l-cysteine (1DCV), a urinary metabolite of trichloroethylene (TCE). Because 1DCV is dichlorinated, only approximately half of the total potential signal is quantifiable when the monoisotopic ion transition (i.e., m/z 256 → 127 for 35Cl2) is monitored. By summing the intensity of a separate and high-abundance 1DCV isotopologue ion transition (i.e., m/z 258 → 129 to include 35Cl and 37Cl), overall signal intensity increased by over 70%. This summation technique improved the analytical sensitivity and limit of detection (LOD) by factors of 2.3 and 2.9, respectively, compared to monitoring the two transitions separately, without summation. Separation and detection were performed using liquid chromatography–tandem mass spectrometry (LC-MS/MS) in negative-ion mode with scheduled selected reaction monitoring. This approach was verified for accuracy and precision using two quality control materials. In addition, we derived a modified signal summation equation to calculate predicted signal enhancements specific to the MITSI approach.

.
Similar content being viewed by others

Introduction
Signal summing is a useful tool for enhancing the signal-to-noise ratio (S/N) and is similar to established signal enhancement and noise reduction techniques, such as bandwidth filtering, ensemble signal averaging, and boxcar averaging [1]. It can be applied to any measurable and repeatable physical response [1]. Its applications include electrocardiography [2, 3], pulse oximetry [4, 5], spectroscopy (nuclear magnetic resonance, Fourier transformations [6]), and digital signal processing (imaging, charge-coupled device binning [7]). Signal summing is based on the principle that multiple signals can commutatively combine given that they are consistent with replicate measurements (i.e., direct current (dc) waveforms), resulting in a precise sum [8, 9]. Moreover, it relies on the resultant noise being random and unrelated to response, such that constructive and deconstructive noise contributions can be made to the overall signal, similar to in-phase and out-of-phase sinusoidal waves (i.e., alternative current (ac) waveforms) [8]. Therefore, the measured signal is expected to improve more rapidly than the noise; quantitatively, the summation of n replicate measurements can improve S/N by a factor of \( \sqrt{n} \) [9].
Mass spectrometry-based quantitative analyses may benefit from signal summing due to the modern mass spectrometer’s ability to precisely measure many signals over short time periods. By coupling a chromatography system and summing the signal intensities of specific ion transitions at given retention times, it becomes easier to differentiate between signal and noise. However, a mass spectrometer can only generate a definite number of data points across a chromatographic peak’s width. The number of data points acquired across a peak is based on dwell and delay times, and is optimized such that quantitation remains accurate and precise (usually ten to fifteen data points) [10]. This restricts the number of chromatographic peak signals that can be summed to improve S/N [11].
Signal summing has been implemented in several recent liquid chromatography–tandem mass spectrometry (LC-MS/MS)-based applications. Nitin et al. demonstrated one of the first instances of selected reaction monitoring (SRM) signal summation [12]. They reported a twofold improvement in the analytical sensitivity of a potential antimalarial agent, bulaquine, and its major metabolite, primaquine, in monkey plasma by summing the two most intense molecular ion transitions for each compound. Swamy et al. demonstrated a more than twofold improvement in analytical sensitivity of two model substrates, clopidogrel and ramiprilat, by summing two distinct SRM ion transition pairs of the same compounds [13]. They reported comparative accuracy and precision between summed and individual approaches. Pauwels et al. applied conventional signal summing to LC-MS/MS detection of the immunosuppressant everolimus by summing several replicate measurements of a single ion transition [11]. They improved the limit of quantitation (LOQ) of everolimus in whole blood from 1.3 to 0.6 μg/L; the resulting S/N improvements were correlated with the \( \sqrt{n} \) relationship. While summing more than three identical transitions was detrimental to the quality of chromatographic peak shape, precision was unaffected. Li et al. implemented signal summation of five identical SRM transitions in a catecholamine assay to achieve sensitivity in the sub-nanogram per liter range [14]. All of the aforementioned studies perform signal summing in one of two ways. The first is to monitor and sum replicates of the exact same ion transition (e.g., summing a ➔ b and a ➔ b). The second is to monitor and sum distinct ion transitions that have the same precursor m/z but different product m/z (e.g., summing a ➔ b and a ➔ c).
Multiple ion transition summation of isotopologues (MITSI) is a novel application of conventional mass spectrometry-based signal summation techniques. MITSI presents a third method of signal summing (e.g., a ➔ b and c ➔ d, where a and c are isotopologue transitions) in addition to the two methods mentioned earlier. This case study examines N-acetyl-S-(1,2-dichlorovinyl)-l-cysteine (1DCV) as a model compound for MITSI and monitors the most abundant ion transitions of two isotopologues of 1DCV in SRM mode. Isotopologues are molecules that have different isotopic compositions (i.e., 35Cl-containing 1DCV and 37Cl-containing 1DCV) [15]. Based on the number of isotopic substitutions in an isotopologue and their relative abundances, the same molecular species can have different molecular masses. Although previous studies have used signal summation of multiple isotopologues, they have been mostly in the context of investigating the dechlorination mechanism of electron ionization [16], or utilizing low-abundance isotopes, such as the determination of the 13C/12C ratio using isotope ratio mass spectrometry [17].
1DCV is a nephrotoxic and mutagenic mercapturic acid [18]. It is a metabolite of trichloroethylene (TCE). Following detoxification via glutathione conjugation, TCE is converted to 1DCV and subsequently excreted in urine [18]. TCE is mostly used in the production of chlorinated chemicals [19]. It is classified as a Group 1 carcinogen by the International Agency for Research on Cancer (IARC) [19,20,21]. Approximately 11 million pounds of TCE was released in the USA between 1998 and 2001. There are community concerns over volatile organic compound exposure, especially for chronic exposures at low concentrations [22, 23].
Several GC-MS methods have been developed for measurement of TCE in blood, tissue, and water [24,25,26,27]. Several LC-MS methods have been developed for measurement of urinary 1DCV [28, 29]. Limits of detection (LODs) for blood and urine assays have been reported as low as 0.01 μg/L [27] and 5.8 μg/L [29], respectively. Despite TCE’s ubiquity, 88% and 99% of samples reported in recent nationally representative US population biomonitoring studies were below the LOD for TCE in blood and 1DCV in urine, respectively [21, 30].
Achieving improvements in analytical sensitivity (and resulting method detection limits) is expensive, as it usually requires upgrades to state-of-the-art instrumentation and a lengthy method development timeframe [31]. We propose MITSI as an inexpensive and simple, yet versatile, methodology that can be used to improve the analytical sensitivity of 1DCV and other high-abundance isotopologues. Details on the theorized benefits of MITSI as compared with other signal summation approaches can be found in Supporting Information (Supp. Info. 1).
Experimental
Materials and Methods
LC-MS Optima-grade acetonitrile was purchased from Fisher Scientific (Suwanee, GA). LC-MS-grade ammonium acetate was purchased from Sigma-Aldrich (St. Louis, MO). Analytical standard-grade 1DCV and 1DCV-[2H3, 13C] were purchased from Toronto Research Chemicals (Ontario, Canada). A human urine pool was collected following a protocol approved by the Centers for Disease Control and Prevention Human Subjects Research Protection Office.
Individual master stocks of unlabeled standard and labeled internal standard were prepared in methanol. Master stocks were diluted in water to make working stocks and stored at − 70 °C prior to use. All calibration solutions were prepared by diluting the working stock solution in 15 mM aqueous ammonium acetate solution. A set of seven calibrators spanning three orders of magnitude in 1DCV concentration was prepared using stable isotope-labeled internal standard (1DCV-[2H3, 13C]).
Samples were prepared following our published method [28]. Briefly, a 50-μL aliquot of sample was mixed with 25 μL of internal standard and 425 μL of 15 mM aqueous ammonium acetate.
LC-MS/MS Analysis
We used an Acquity UPLC Classic system equipped with a 2.1 × 150 mm, 1.8 μm HSS T3 C18 column (Waters Corporation, Milford, MA). The UPLC system was coupled to a Triple Quad 5500 mass spectrometer equipped with an electrospray ionization (ESI) source (SCIEX, Framingham, MA). Chemical separation was performed using a solvent gradient of 15 mM aqueous ammonium acetate (mobile phase A) and acetonitrile (mobile phase B) [28]. Column and sample manager temperatures were set to 40 °C and 25 °C, respectively. The injection volume was 2 μL using full loop injection mode. The mass spectrometer was operated in negative-ion ESI scheduled SRM mode. Ion source parameters were optimized as follows: ESI voltage, − 4500 V; CAD gas, 7 psi; curtain gas flow, 35 psi; nebulizing gas (GS1) flow, 45 psi; heating gas (GS2) flow, 55 psi; and heater temperature, 650 °C. Compound-specific mass spectrometric parameters for 1DCV and 1DCV-[2H3, 13C] were optimized for each high-abundance ion transition (Supp. Info. 2, Supp. Table 1). All transitions were investigated for potential interferences or matrix effects prior to implementation.
Data Analysis
All LC-MS/MS data were generated in Analyst 1.6 (SCIEX, Framingham, MA) and processed in MultiQuant 3.0.3 (SCIEX, Framingham, MA), including signal summation. Calibration curves were fitted using a linear regression of 1/x weighted peak area data with the coefficient of determination exceeding 0.99 for both SRM transitions. The signal intensities generated separately for each ion transition were summed to create the SUM transition, post-acquisition (Supp. Info. 3).
Results and Discussion
Isotopic Distribution and MITSI Application to 1DCV
1DCV is a dichlorinated compound with molecular formula C7H9Cl2NO3S. Both 35Cl and 37Cl isotopes are in high natural abundance, giving 1DCV a proportionally high ratio of the 35Cl-containing 1DCV isotopologue compared with the 37Cl-containing 1DCV isotopologue. 1DCV’s structure and empirical and theoretical mass distributions are shown in Figure 1. The distributions show that the most abundant monoisotopic m/z of 1DCV in negative-ion mode contains two 35Cl atoms (m/z 256, relative intensity of 100%). The second most abundant m/z contains one 35Cl and one 37Cl atom (m/z 258, relative intensity of 70% with respect to m/z 256). Additional m/z values are also present at varying, but lesser, abundances (e.g., two 37Cl atoms). If the major isotopologue at m/z 256 were to be the only ion transition monitored, it would account for approximately 49% of total possible signal intensity. Accordingly, there is approximately 51% of total signal intensity consisting of individually less abundant m/z values; thus, there is an opportunity for a near doubling of signal intensity if all ion transitions of 1DCV isotopologues are measured and summed. However, monitoring only the next most abundant ion transition at m/z 258 yields signal gains of 70%, our preferred approach. In this case, MITSI was used to sum two isotopologue ion transitions, SRM1 (m/z 256 → 127) and SRM2 (m/z 258 → 129), to increase S/N, and thus sensitivity.

Experimental (blue) and predicted (red) isotopic mass distributions of 1DCV, with the molecular structure shown on the inset
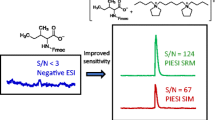
The chromatograms generated using SRM1 and SRM2, as well as their summed peak (referred to as SUM), are depicted in Figure 2. Both SRMs produced near identical signal responses for 6.2 μg/L 1DCV in 15 mM aqueous ammonium acetate, with response differences matching the isotopologue abundances shown in Figure 1. MITSI increased the signal intensity of SUM by approximately 75% with respect to SRM1 and 130% with respect to SRM2.

Overlay of 1DCV chromatograms using ion transitions: SRM1 (m/z 256 → 127), SRM2 (m/z 258 → 129), and SUM (summation of SRMs 1 and 2)
Analytical figures of merit were evaluated for the individual and summed ion transitions using a set of seven calibrators across three orders of magnitude in 1DCV concentration. The calculated slopes of the 1/x weighted linear regressions for SRM1 and SRM2 peak area calibration curves were 0.029 and 0.022, respectively (Figure 3). SRM1 is expected to have a greater slope than SRM2 due to its higher signal intensity. In addition, LODs and LOQs were calculated using spiked urine samples at four different concentrations following Taylor’s method [32]. The standard deviation at zero concentration, S0, was extrapolated by plotting standard deviation versus calculated concentration (Figure 4). The LODs and LOQs were calculated as 3S0 and 10S0, respectively. The LODs calculated using SRM1 and SRM2 were 37.5 and 57.9 ng/L, respectively, and the LOQs were 125 and 193 ng/L, respectively (Table 1).

Linear regression fit of calibration curves for SRM1 (m/z 256 → 127), SRM2 (m/z 258 → 129), and SUM (summation of SRMs 1 and 2)

Standard deviation versus concentration of triplicate injections for the calculation of LODs and LOQs of 1DCV
Following signal summation, the SUM calibration curve was plotted using another linear regression of 1/x weighted peak area (Figure 3). The calculated slope of the SUM calibration curve was 0.051. This was 75% and 132% greater than the curves generated using SRM1 and SRM2, respectively. Thus, the signal intensity of 1DCV using the signal summing approach was 1.8 and 2.3 times greater than SRM1 and SRM2, respectively. This is due to the percent increases in peak area of SUM relative to SRM1 (75%) and SRM2 (130%), coupled with decreasing noise. Similarly, the calculated LOD and LOQ for SUM were 20.1 ng/L and 67.0 ng/L, respectively. These values were 1.9 and 2.9 times lower than SRM1 and SRM2, respectively (Table 1). This noteworthy improvement of LOD and LOQ is attributed to the improvement in the method precision that MITSI affords by enhancing signal and diminishing noise.
In addition, verification of the accuracy and precision of the MITSI approach was studied using quality control (QC) materials. Two QC materials (QC-1 and QC-2) were prepared in-house by spiking known amounts of 1DCV in a human urine pool. Two sets of each QC sample, prepared fresh from stock each day, were run over 10 weeks on three different instruments, resulting in 20 total results for each QC. The calculated concentrations for SUM, SRM1, and SRM2 (Table 2) were within 2.1% accuracy and 5.8% relative standard deviation (RSD), demonstrating that the MITSI approach does not compromise accuracy. We observed directional improvement in the method precision in the lower concentration region, as shown by a 1.4% improvement in RSD at the QC-1 concentration range. Less pronounced improvement in the precision was observed in the LOQ concentration range, as shown by a maximum of 0.5% reduction in RSD at the QC-2 concentration range with respect to SRM1. The magnitude of this directional improvement in RSD is hypothesized to be concentration related. Higher concentrations result in greater signal intensity and are less affected by random fluctuations in baseline noise. Alternatively, lower concentrations, and hence lower signal intensities, are relatively more sensitive to noise variation. This results in greater improvements in RSD when summing at low concentrations (i.e., smaller RSDs post-summing) while maintaining linear response (Figure 3).
QC verification demonstrates that MITSI can be employed without sacrificing method accuracy or introducing laborious method development activities. Through these multiple QC analyses, we show that MITSI improves precision and LODs compared with the original ion transitions, while maintaining accuracy.
Application of the MITSI-Modified \( \sqrt{n} \) Proportion
It is essential to investigate the applicability of the theorized \( \sqrt{n} \) increase in S/N when summing n signals and how this may apply to MITSI. The \( \sqrt{n} \) proportion has been derived in literature and can be found in Supp. Info. 4. It is summarized in Eq. (1) [33], where n is the number of signals summed, Si is the individual signal, and (σN2) is the additive variance of the signals.
However, MITSI does not sum replicate measurements and thus Eq. (1) must be modified to Eq. (2). The derivation is shown in Supp. Info. 4, where SR is the signal of a relative ion transition and Ai is the relative abundance of the ion transitions to be summed.
Based on Eq. (1), a 41% increase in S/N between the summed transitions and the individual transitions is expected when two replicate signals are summed. Alternatively, based on Eq. (2), two separate increases are predicted, based on which ion transition is deemed the relative transition, SR. If SRM1 is the relative transition with relative intensity of 100% (or 1.0), SRM2 then has relative intensity of 70% (or 0.7) based on 1DCV isotopologue abundance. Assuming n = 2, this results in a projected S/N increase of \( \kern0.5em \frac{\left(1.00+0.70\right)}{\sqrt{2}}\bullet \frac{S_{\mathrm{R}}}{\sigma_N} \), which simplifies to 1.21, or a 21% increase in S/N. However, this percentage only states the relative increase of S/N between SUM:SRM1. To obtain the relative increase of SUM:SRM2, we modify SRM2 to be the relative transition, SR. If SRM2’s relative signal is 100%, SRM1’s relative signal is 130%, or 1.3. Accordingly, the projected S/N increase of SUM:SRM2 is \( \frac{\left(1.00+1.30\right)}{\sqrt{2}}\bullet \frac{S_{\mathrm{R}}}{\sigma_N} \), which simplifies to 1.64, or a 64% increase. These S/N increases should be more accurate projections than 1.41, or a 41% increase, which is based solely on the number (and not the relative intensity) of the signals. The projected 41% increase would only be accurate if we summed replicate transitions of the same intensity.
To further explore Eq. (2)’s utility for predicting accurate signal enhancements, experimentally calculated peak areas (Supp. Info. 2, Supp. Table 2) can be used as an estimative proxy for signal abundance. By substituting experimentally determined average peak area increases (Supp. Info. 2, Supp. Table 3), we can calculate an empirical version of expected sensitivity increase. Doing so yields projected increases of 27% \( \left(\frac{\left(1.00+0.79\right)}{\sqrt{2}}=1.27\right) \) and 60% \( \left(\frac{\left(1.00+1.26\right)}{\sqrt{2}}=1.60\right) \) for SUM:SRM1 and SUM:SRM2 S/N values, respectively. These are in agreement with the 21% and 64% predicted increases estimated in the previous paragraph.
Paradox of Predicted and Experimental Signal-To-Noise Enhancements
The validity of the \( \sqrt{n} \) proportion can be tested by comparing experimentally calculated S/N improvements in QC-1 and QC-2 with and without using MITSI. Experimental S/N values were calculated using three different available integration algorithms within MultiQuant software: MQ4, Summation, and SignalFinder (Supp. Info. 2, Supp. Table 4). Several parameters that affect the S/N value calculated within the software can be changed (Supp. Info. 2, Supp. Table 5).
No conclusive evidence of the \( \sqrt{n} \) proportion was found when comparing the S/Ns computed within the MultiQuant software with those predicted by Eq. (2). This result is contrary to the agreement of Eq. (2) with peak area increases shown earlier. These values are reported (Supp. Info. 2, Supp. Table 6) as percent enhancement factors (% EFs), calculated as the percent quotient of the S/N of the SUM and either SRM1 or SRM2, based on Swamy et al. [13].
The calculated % EFs using MultiQuant S/N values differ from the predicted % EFs. This is hypothesized to result from two factors: complexity of mass spectrometer noise and variations in defining S/N. Both of these factors are expected to cause deviations from the \( \sqrt{n} \) proportionality [8]. First, if all noise were Gaussian, then SUM would approach a noise-free signal, since noise variance is additive [33]. However, our signal (as in all mass spectrometers) is subject to non-random, non-canceling external noise from atmospheric fluctuations and instrument drift [34]. Second, MultiQuant uses a “relative noise” model to calculate S/N. Our assumptions in deriving Eqs. (1) and (2) rely on S/N equating to the inverse of RSD, but MultiQuant does not define its S/N outputs as such [35]. Thus, there are quantitation differences between expected S/N (based on Eqs. (1) and (2)) and calculated S/N (based on MultiQuant outputs).
Interestingly, peak area averages (Supp. Info. 2, Supp. Table 2) show little variation among the three integration algorithms, while S/N values calculated by MultiQuant (Supp. Info. 2, Supp. Table 6) vary widely. This is because the integration algorithms calculate peak areas in similar manners, but calculate relative noise differently [35]. This explains why signal response is consistent across integration algorithms, while S/N is not.
The subjectivity of the S/N calculations is a hindrance for predicting quantifiable MITSI signal enhancements. Surprisingly, peak area increases may be more indicative of potential MITSI benefits until more applicable noise calculations can be developed. There is a clear need for more accurate and precise S/N calculations, but due to algorithmic variance and the many sources of noise in LC MS/MS, this is difficult to achieve. These difficulties may explain why few signal summing papers have attempted to correlate the \( \sqrt{n} \) proportion to their results. Until a more accurate predictive model becomes available, peak area increases and Eq. (2) (along with comparisons of accuracy, precision, LODs, and LOQs) are the best ways to predict MITSI’s potential workflow benefits.
Conclusions
Multiple ion transition summation of isotopologues (MITSI) is a useful approach to improve the analytical sensitivity in mass spectrometric detection of compounds containing two or more high-abundance isotopes. It offers additional benefits to conventional signal summation techniques by taking advantage of analytes with multiple abundant natural isotopic substitutions. We applied MITSI to enhance the detection of 1DCV, a metabolite of trichloroethylene. By summing signal intensities across the peak of the same analyte, but two different molecular ion transitions, signal intensity was enhanced up to 130%. Corresponding ease of integration and reductions in baseline noise improved method precision and lowered both the LOD and LOQ by up to threefold. We verified the method for accuracy and precision using two quality control materials; it is a viable approach to improve analytical sensitivity of high-throughput LC-MS/MS analyses of chlorinated compounds. In addition, a modified S/N enhancement equation specific to MITSI was derived and proposed as an accurate tool for measuring potential peak area enhancements, as opposed to more subjective S/N calculations.
The novel MITSI methodology proposed here has promising applications to additional chlorinated compounds of interest (e.g., 2DCV, a constitutional isomer of 1DCV; N-acetyl-S-(trichlorovinyl)-l-cysteine, a metabolite of tetrachloroethylene). Other potential applications include halogenated compounds such as dioxins, polychlorinated biphenyls, and halocarbons (e.g., organobromines). These classes of compounds are of significant public health interest, as they are present at low levels in the environment [36,37,38]. Although the applications of signal summing are wide reaching, it is an underutilized technique in quantitative mass spectrometry.
Change history
31 May 2019
The original article has been corrected to include the missing chemical structure in Fig. 1.
References
Hieftje, G.M.: Signal-to-noise enhancement through instrumental techniques. Part II. Signal averaging, boxcar integration, and correlation techniques. Anal. Chem. 44, 69A–78A (1972)
Dobrev, D.: Two-electrode low supply voltage electrocardiogram signal amplifier. Med. Biol. Eng. Comput. 42, 272–276 (2004)
Kelsey, R.M., Guethlein, W.: An evaluation of the ensemble averaged impedance cardiogram. Psychophysiology. 27, 24–33 (1990)
Bindszus, A., Boos, A.: Pulse Rate and Heart Rate Coincidence Detection for Pulse Oximetry. US patent No. 6178343 B1, January 23 (2001)
Baker, C.R., Jr.: Selection of Ensemble Averaging Weights for a Pulse Oximeter Based on Signal Quality Metrics. US patent No. 7194293 B2, March 20 (2007)
Clark, W., Hanson, M., Lefloch, F.: Magnetic resonance spectral reconstruction using frequency-shifted and summed Fourier transform processing. Rev. Sci. Instrum. 66, 2453–2464 (1998)
Epperson, P.M., Denton, M.B.: Binning spectral images in a charge-coupled device. Anal. Chem. 61, 1513–1519 (1989)
Hieftje, G.M.: Signal-to-noise enhancement through instrumental techniques: part I. Signals, noise, and S/N enhancement in the frequency domain. Anal. Chem. 44, 81A–88A (1972)
Trimble, C.: What is signal averaging? Hewlett-Packard Journal. 19, 2–16 (1968)
SCIEX. The scheduled MRM™ algorithm enables intelligent use of retention time during multiple reaction monitoring. https://sciex.com/Documents/Downloads/Literature/mass-spectrometry-ScheduledMRM-0921010.pdf. Accessed July 2018
Pauwels, S., Jans, I., Peersman, N., Billen, J., Vanderschueren, D., Desmet, K., Vermeersch, P.: Possibilities and limitations of signal summing for an immunosuppressant LC-MS/MS method. Anal. and Bioanal. Chem. 407, 6191–6199 (2015)
Nitin, M., Rajanikanth, M., Lal, J., Madhusudanan, K.P., Gupta, R.C.: Liquid chromatography–tandem mass spectrometric assay with a novel method of quantitation for the simultaneous determination of bulaquine and its metabolite, primaquine, in monkey plasma. J. Chromatogr. B. 793, 253–263 (2003)
Swamy, J., Kamath, N., Shekar, A., Srinivas, N., Kristjansson, F.: Sensitivity enhancement and matrix effect evaluation during summation of multiple transition pairs—case studies of clopidogrel and ramiprilat. Biomed. Chromatogr. 24, 528–534 (2010)
Li, X., Li, S., Kellermann, G.: An integrated liquid chromatography–tandem mass spectrometry approach for the ultra-sensitive determination of catecholamines in human peripheral blood mononuclear cells to assess neural-immune communication. J. Chromatogr. A. 1449, 54–61 (2016)
Schwarzenbach, R., Gschwend, P., Imboden, D.: Environmental Organic Chemistry, 3rd edn. John Wiley & Sons, Inc., Hoboken, N.J (2016)
Tang, C., Tan, J. (2017). Observation of Concerted and Stepwise Multiple Dechlorination Reactions of Perchlorethylene in Electron Ionization Mass Spectrometry According to Measured Chlorine Isotope Effects. arXiv preprint arXiv:1709.01739. Retrieved October 2018 from the arXiv database.https://arxiv.org/abs/1709.01739
Zhang, Y., Tobias, H.J., Sacks, G.L., Brenna, J.T.: Calibration and data processing in gas chromatography combustion isotope ratio mass spectrometry. Drug Test. Anal. 4, 912–922 (2012)
Birner, G., Vamvakas, S., Dekant, W., Henschler, D.: Nephrotoxic and genotoxic N-acetyl-S-dichlorovinyl-L-cysteine is a urinary metabolite after occupational 1,1,2-trichloroethene exposure in humans: implications for the risk of trichloroethene exposure. Environ. Health Perspect. 99, 281–284 (1993)
Guha, N., Loomis, D., Grosse, Y., Lauby-Secretan, B., Ghissassi, F.E., Bouvard, V., Benbrahim-Tallaa, L., Baan, R., Mattock, H., Straif, K.: Carcinogenicity of trichloroethylene, tetrachloroethylene, some other chlorinated solvents, and their metabolites. The Lancet Oncology. 13, 1192–1193 (2012)
Jollow, D.J., Bruckner, J.V., McMillan, D.C., Fisher, J.W., Hoel, D.G., Mohr, L.C.: Trichloroethylene risk assessment: a review and commentary. Crit. Rev. Toxicol. 39, 782–797 (2009)
IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Trichloroethylene, tetrachloroethylene, and some other chlorinated agents. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, No. 106, (2014) Lyon:International Agency for Research on Cancer
Gostner, J.M., Zeisler, J., Alam, M.T., Gruber, P., Fuchs, D., Becker, K., Neubert, K., Kleinhappl, M., Martini, S., Überall, F.: Cellular reactions to long-term volatile organic compound (VOC) exposures. Sci. Rep. 6, 37842 (2016)
Bernstein, J.A., Alexis, N., Bacchus, H., Bernstein, I.L., Fritz, P., Horner, E., Li, N., Mason, S., Nel, A., Oullette, J., Reijula, K., Reponen, T., Seltzer, J., Smith, A., Tarlo, S.M.: The health effects of nonindustrial indoor air pollution. J. Allergy Clin. Immunol. 121, 585–591 (2008)
Brown, S.D., Dixon, A.M., Bruckner, J.V., Bartlett, M.G.: A validated GC-MS assay for the quantitation of trichloroethylene (TCE) from drinking water. Int. J. Environ. Anal. Chem. 83, 427–432 (2003)
Dixon, A.M., Brown, S.D., Muralidhara, S., Bruckner, J.V., Bartlett, M.G.: Optimization of SPME for analysis of trichloroethylene in rat blood and tissues by SPME-GC/MS. Instrumentation Science & Technology. 33, 175–186 (2005)
Liu, Y., Muralidhara, S., Bruckner, J.V., Bartlett, M.G.: Determination of trichloroethylene in biological samples by headspace solid-phase microextraction gas chromatography/mass spectrometry. J. Chromatogr. B. 863, 26–35 (2008)
Blount, B.C., Kobelski, R.J., McElprang, D.O., Ashley, D.L., Morrow, J.C., Chambers, D.M., Cardinali, F.L.: Quantification of 31 volatile organic compounds in whole blood using solid-phase microextraction and gas chromatography–mass spectrometry. J. Chromatogr. B. 832, 292–301 (2006)
Alwis, K.U., Blount, B.C., Britt, A.S., Patel, D., Ashley, D.L.: Simultaneous analysis of 28 urinary VOC metabolites using ultra high performance liquid chromatography coupled with electrospray ionization tandem mass spectrometry (UPLC-ESI/MSMS). Anal. Chim. Acta. 750, 152–160 (2012)
Suh, J.H., Eom, H.Y., Kim, U., Kim, J., Cho, H.-D., Kang, W., Kim, D.S., Han, S.B.: Highly sensitive electromembrane extraction for the determination of volatile organic compound metabolites in dried urine spot. J. Chromatogr. A. 1416, 1–9 (2015)
Centers for Disease Control and Prevention. National Health and Nutrition Examination Survey. https://wwwn.cdc.gov/nchs/nhanes/2011-2012/UVOC_G.htm. Accessed September 2018
Grebe, S.K., Singh, R.J.: LC-MS/MS in the clinical laboratory - where to from here? The clinical biochemist. Reviews. 32, 5–31 (2011)
Taylor, J.: Quality Assurance of Chemical Measurements. Lewis Publishers, New York (1987)
Hassan, U., Anwar, M.: Reducing noise by repetition: introduction to signal averaging. Eur. J. Phys. 31, 453–465 (2010)
Marshall, A.G., Verdun, F.R.: Fourier Transforms in NMR, Optical, and Mass Spectrometry: a User’s Handbook. Elsevier, Amsterdam, The Netherlands (1990)
SCIEX. MultiQuant software reference guide. https://sciex.com/documents/community/support/mq-glp-software-reference-guide.pdf. Accessed October 2018
Reggiani, G., Bruppacher, R.: Symptoms, signs and findings in humans exposed to PCBs and their derivatives. Environ. Health Perspect. 60, 225–232 (1985)
Mizouchi, S., Ichiba, M., Takigami, H., Kajiwara, N., Takamuku, T., Miyajima, T., Kodama, H., Someya, T., Ueno, D.: Exposure assessment of organophosphorus and organobromine flame retardants via indoor dust from elementary schools and domestic houses. Chemosphere. 123, 17–25 (2015)
Haug, L.S., Sakhi, A.K., Cequier, E., Casas, M., Maitre, L., Basagana, X., Andrusaityte, S., Chalkiadaki, G., Chatzi, L., Coen, M., de Bont, J., Dedele, A., Ferrand, J., Grazuleviciene, R., Gonzalez, J.R., Gutzkow, K.B., Keun, H., McEachan, R., Meltzer, H.M., Petraviciene, I., Robinson, O., Saulnier, P.-J., Slama, R., Sunyer, J., Urquiza, J., Vafeiadi, M., Wright, J., Vrijheid, M., Thomsen, C.: In-utero and childhood chemical exposome in six European mother-child cohorts. Environ. Int. 121, 751–763 (2018)
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention. Use of trade names is for identification purposes only and does not imply endorsement by the Centers for Disease Control and Prevention, the Public Health Service, or the U.S. Department of Health and Human Services.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
The original version of this article was revised: it was corrected to include the missing chemical structure in Figure 1.
Electronic supplementary material
ESM 1
(DOCX 1071 kb)
Rights and permissions
About this article

Cite this article
Movassaghi, C.S., McCarthy, D.P., Bhandari, D. et al. Multiple Ion Transition Summation of Isotopologues for Improved Mass Spectrometric Detection of N-Acetyl-S-(1,2-dichlorovinyl)-L-cysteine. J. Am. Soc. Mass Spectrom. 30, 1213–1219 (2019). https://doi.org/10.1007/s13361-019-02169-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-019-02169-8