
Abstract
DraIII is a type IIP restriction endonucleases (REases) that recognizes and creates a double strand break within the gapped palindromic sequence CAC↑NNN↓GTG of double-stranded DNA (↑ indicates nicking on the bottom strand; ↓ indicates nicking on the top strand). However, wild type DraIII shows significant star activity. In this study, it was found that the prominent star site is CAT↑GTT↓GTG, consisting of a star 5′ half (CAT) and a canonical 3′ half (GTG). DraIII nicks the 3′ canonical half site at a faster rate than the 5′ star half site, in contrast to the similar rate with the canonical full site. The crystal structure of the DraIII protein was solved. It indicated, as supported by mutagenesis, that DraIII possesses a ββα-metal HNH active site. The structure revealed extensive intra-molecular interactions between the N-terminal domain and the C-terminal domain containing the HNH active site. Disruptions of these interactions through site-directed mutagenesis drastically increased cleavage fidelity. The understanding of fidelity mechanisms will enable generation of high fidelity REases.
Similar content being viewed by others


Introduction
Restriction endonucleases (REases) are components of restriction-modification systems that occur ubiquitously among prokaryotic organisms and are among the basic tools of molecular biology (Roberts, 2005). To date, more than 3600 REases representing more than 250 different specificities have been characterized and classified into four types (Roberts et al., 2003). Type II REases are widely used in genetic technologies because of their stable cleavage pattern and are the most well studied. They recognize short DNA sequences (3–8 bp) and create double strand breaks at constant positions within or close to that sequence to generate new ends with 5′-phosphates and 3′-hydroxyls (Roberts et al., 2003). In particular, type IIP REases recognize palindromic sequences and create symmetrical double strand breaks.
Although REases usually cleave specific DNA sequences accurately, certain REases cleave sequences which are similar, but not identical, to their defined recognition sequences under non-optimal conditions, such as the presence of organic solvent, sub-optimal pH values and high enzyme concentrations. This relaxed specificity has been termed star activity. Star activity has been reported with REases such as XbaI, SalI, PstI, BamHI, PvuII and EcoRV (Malyguine et al., 1980; Nasri and Thomas, 1987; Robinson and Sligar, 1995). Star activity is not desirable for most REase applications where off-site cleavage is detrimental. Because star activity is relatively weak, the star sites of only a few enzymes have been defined. Interestingly, all identified star sites are only different from the canonical sequence by one base (George and Chirikjian, 1982; Halford et al., 1986; Nasri and Thomas, 1987). Divalent ions and reaction conditions, such as pH, salt concentration and neutral detergents, can enhance or temper the fidelity of some REases (Robinson and Sligar, 1995; Saravanan et al., 2007b). REases of high fidelity have been developed and are commercially available (High Fidelity Restriction Endonucleases, United States Patent No.: US8372619B2, New England Biolabs). The mechanism by which the fidelity increases is still unclear (Wei et al., 2008).
Compared to the fidelity determination mechanism, the catalytic mechanisms of type II REases are well characterized. Several distinct catalytic site motifs and mechanisms have been identified among type II REases (Orlowski and Bujnicki, 2008). The most common catalytic motif, the PD-(D/E)XK superfamily, has evolved to recognize short DNA sequences with the catalytic residues surrounded by a densely packed array of side chains that can fit and interact with the target bases in the major groove (Horton et al., 2004; Orlowski and Bujnicki, 2008). Alternative catalytic motifs, associated with different core protein folds have been experimentally identified in many type II REases. These include the HNH motif in KpnI, MnlI, PacI, Eco31I, Hpy99I and HpyAV (Chan et al., 2010; Jakubauskas et al., 2007; Kriukiene, 2006; Saravanan et al., 2004; Shen et al., 2010; Sokolowska et al., 2009), the GIY-YIG motif in Hpy188I and Eco29kI (Ibryashkina et al., 2007; Kaminska et al., 2008), the phospholipase D motif in BfiI (Sapranauskas et al., 2000) and a “half pipe” fold in PabI (Miyazono et al., 2007). A comprehensive bioinformatics study has predicted numerous new members of each of these sub-groups, especially the HNH REases (Orlowski and Bujnicki, 2008).
The conserved structural element of the HNH motif is known as the ββα-metal fold because of its two anti-parallel β-strands connected by a loop of variable length and the α-helix that follows (Mehta et al., 2004). The binding site for a single catalytic divalent metal ion, typically magnesium, is embedded within this fold. To date the co-crystal structures of Hpy99I and PacI with their canonical DNA are the only atomic structures that show the intricate protein-DNA interactions of HNH type IIP REases (Shen et al., 2010; Sokolowska et al., 2009). The substrate DNA is severely bent in both structures due to the expansion of the minor groove. In PacI, the DNA distortion is accompanied by unstacked bases and non-canonical A:A and T:T base pairs. This is a clear divergence from the invariant bending of the substrate DNA through the expansion of the major groove by the PD-(D/E)XK REases (Horton et al., 2004).
DraIII is a type IIP REase isolated from Deinococcus radiophilus ATCC 27603. It recognizes the gapped double-stranded DNA sequence CAC↑NNN↓GTG (↑ indicates nicking on the bottom strand; ↓ indicates nicking on the top strand) and nicks at 5′ of the first G on both strands, thus creating a double strand break with a three base 3′ overhang (Grosskopf et al., 1985). DraIII exhibits significant star activity in the presence of Mg2+. Considering its star activity and small size (25.7 kDa), we used DraIII as a model system to investigate the determinants of cleavage fidelity of REases.
RESULTS
Determination of DraIII Fidelity Index (FI)
A quantitative definition of star activity is provided by the Fidelity Index (FI), which is the ratio of the highest quantity of a REase showing no star activity during digestion to the lowest quantity needed for complete digestion on canonical sites on a specific substrate DNA (Wei et al., 2008). A higher FI corresponds to a higher cleavage fidelity of the REase. DraIII has an FI of 2 on λ DNA under reaction conditions described in MATERIALS AND METHODS (Fig. 1A).

Determination ofDraIII Fidelity Index (FI) and star sites. (A) Determination of DraIII Fidelity Index (FI). λ DNA (1.6 nmol/L; 16 nmol/L CACNNNGTG sites) is digested by DraIII in a series of two fold dilutions. DraIII concentration: Lane 1, 3.2 μmol/L; Lane 10, 6.25 nmol/L; Lane 11, 3.125 nmol/L; Lane 21, 3.05 pmol/L; Lane 22, 1-kb DNA Ladder (NEB). The vertical arrows indicate the two critical points: HNS—the Highest REases concentration showing No Star activity and LCC—the Lowest REase concentration needed for Complete Cleavage on canonical sites. FI = HNS/LCC, which is 2 in this case. Asterisk represents a star band, and the hash represents a band that resulted from partial cleavage of λ DNA. The theoretical digestion pattern of DraIII to λ DNA was predicted using NEBcutter (Vincze et al., 2003) and was shown on the left. (B) DraIII star site in pUC19 was predicted to be the CATGTTGTG site. Lane 1: BamHI (cut at nt 417) and XmnI (cut at nt 2298) double digestion on pUC19 generated the 1.9-kb and 0.8-kb bands. Lane 2: BamHI, XmnI and DraIII triple digestion on pUC19. Asterisk indicates the star band. According to the approximate size of star bands, the CATGTTGTG site (nt 2033) was hypothesized to be the DraIII star site. Cleavage on predicted CATGTTGTG site generated the 1.6-kb and 0.3-kb star bands. Lane 3: 1-kb DNA Ladder. (C) DraIII star activity cleaves the CATGTTGTG site in pXba. Lane 1: 1-kb DNA Ladder. Lane 2: pXba was digested by DraIII. Asterisk indicates the star bands. DraIII star activity generates the expected 6.5-kb and 4.5-kb star bands on pXba. (D) DraIII star activity shows selectivity to the central “NNN” part of CATNNNGTG site. There are 11 CATNNNGTG sites in pXba and the sequences containing these sites were tested independently on oligonucleotide duplex DNAs carrying each of the sites (Table S1). The canonical CACGGCGTG site was used as positive control. DraIII shows cleavage activity to CATATGGTG, CATTACGTG, CATGTGGTG, CATAAAGTG and CATGTTGTG sites. DraIII did not cut the pseudo-palindromic CATGTTATG site
Determination of DraIII star sites
In addition to the expected bands resulting from cleavage of the 10 canonical CACNNNGTG sites, extra discrete bands were observed in the cleavage reactions of λ DNA (Fig. 1A). These bands were attributed to star activity. The defined size of the star bands from cleavage reactions of λ DNA suggests that the DraIII star activity is not random and has certain specificity. It is difficult to map the DraIII star sites in λ DNA because of its large size (48.5-kb) and the large number of possible star sites. A smaller 2.7-kb pUC19 DNA was used to determine DraIII star sites. Although it does not contain the DraIII canonical site, the pUC19 DNA was linearized by DraIII (data not shown), showing the presence of one or more DraIII star sites. Double (BamHI and XmnI) and triple (BamHI, XmnI and DraIII) digestion on pUC19 suggest that DraIII star activity generates two star bands through one cut (Fig. 1B). According to the approximate size of star bands, the CATGTTGTG site (nt 2033) which only differs from the canonical sequence by one base was hypothesized to be DraIII star site. Cleavage on predicted CATGTTGTG star site generated the 1.6-kb and 0.3-kb star bands (Fig. 1B, Lane 2). A larger 22.6-kb plasmid DNA pXba was used to verify the predicted star site. pXba contains three DraIII canonical sites (nt 5816, 16551 and 17404 respectively) and one predicted star site CATGTTGTG (nt 21910). It was found that DraIII generated the 6.5-kb and 4.5-kb star bands consistent with cleavage at the CATGTTGTG site (Fig. 1C, Lane 2).
Since DraIII’s canonical specificity has no preference for the middle three nucleotides (Grosskopf et al., 1985), the specificity of these nucleotides within the star site CATNNNGTG was investigated. There are 11 such sites in pXba and the sequences containing these sites were tested independently on oligonucleotide duplex DNAs carrying each of the sites (Table S1). Among the 11 CATNNNGTG sites (Table S1, DNA2–12), cleavage was observed where NNN = AAA, GTG, TAC, ATG and GTT (Fig. 1D). Apparently the star activity of DraIII has a sequence preference for the central three nucleotides, whereas the canonical cleavage activity does not (Grosskopf et al., 1985).
DraIII digests star site sequence in asymmetrical pattern
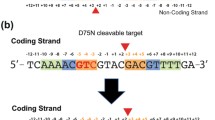
To locate the exact cleavage position within the CATGTTGTG star site, 39-bp duplex DNA which contains canonical or star site and a 5′ Cy5 fluorophore on each strand (Fig. 2A) was digested by DraIII and separated by 20% acrylamide urea PAGE (Fig. 2B). The single strand product was compared with synthesized 15, 16, 17, 25, 26 and 27-nt single strand DNA markers (Fig. 2A and 2B). The PAGE result confirmed that DraIII star site is CAT↑GTT↓GTG (↑ indicates nicking on the bottom strand; ↓ indicates nicking on the top strand) (Fig. 2B).

DraIII digests star site sequence in asymmetrical pattern. (A) Sequences of Cy5-labeled DNA substrates. (B) Star activity cleavage occurs in CAT↑GTT↓GTG. Cy5-labeled canonical or star DNA was digested by DraIII as described in Supplementary EXPERIMENTAL. The single strand product was separated by TBE urea polyacrylmide gel and compared with synthesized single-stranded markers. Star activity cleavage occurs in CAT↑GTT↓GTG (↑ indicates nicking on the bottom strand; ↓ indicates nicking on the top strand). (C and D) DraIII digests a star site sequence in an asymmetrical manner. 100 nmol/L Cy5-labeled star DNA was digested by 500 nmol/L DraIII. The samples were collected at the designated time intervals and were analyzed by electrophoresis using TBE urea polyacrylmide gel. The amount of the quantified products was plotted against time
To decipher the DraIII star activity cleavage mode, 39-bp duplex star DNA was used for time course analysis. Cleavage products were collected at different time points and analyzed by denaturing urea polyacrylamide gel electrophoresis. The 16-nt single strand product (5′-Cy5-catgatccccCATGTT) represents nicking at the top canonical half site and the 26-nt single strand product (5′-Cy5-atggtagagacgcttttttgCACAAC) represents nicking at the bottom star half site (Fig. 2A, star DNA; Fig. 2C). The nicking on the two halves of the DraIII star sequence occurs at different rates: the canonical half site GTG was cleaved faster than the star half site CAT (Fig. 2D). No difference in the cleavage rate of the two half sites for the canonical DraIII site was observed (data not shown). We further tested whether DraIII star activity cleaves the star half site at an equal or slower rate if the star half site CAT is present in both half sites arranged in the pseudo-palindrome (CATGTTATG). DraIII showed no significant activity on this pseudo-palindromic site (Fig. 1D, DNA13). This shows that under star conditions DraIII cleaves the canonical half site faster than the star half site, and that it requires one canonical half site for cleavage to occur. Altering the flanking sequence (Table S1, DNA14) showed no influence on the cleavage of the CATGTTGTG star site (data not shown).
Overall protein structure and catalytic sites
The structure of DraIII was determined in the presence of an 11-bp canonical DNA duplex containing a phosphorothioate at both of the scissile phosphodiester bonds (Fig. 3B). A complete protein structure was derived from a protein-phosphorothioate DNA duplex in the presence of magnesium chloride, although the DNA was not observed in the structure (Fig. 3A and Table 1). DraIII exists as a homodimer and dimerization occurs in the C-terminus region where the ββα-metal HNH active site is located. Two zinc- and one magnesium-binding sites are found in each DraIII monomer (Fig. 3A).

TheDraIII overall protein structure. (A) The complete DraIII structure derived in the presence of magnesium chloride. The ion binding motifs are shown. (B) The 11-bp phosphorothioate canonical DNA duplex. (C) The secondary structure of the DraIII subunit. Elements of secondary structure are indicated by α-helix (red rectangle) and β-strand (blue arrow). Catalytic residues of the HNH endonuclease motif are indicated with blue font. Zinc-binding cysteine residues are indicated with red font. The location of N-terminal domain, C-terminal domain and ββα-metal motif is indicated by the orange, cyan and black underlines respectively
The DraIII monomer contains a ββα-metal fold (β1-β2-α12) in the C-terminal domain where the HNH active site is located (Fig. 3C). Sequence and structure alignments reveal that amino acid residue N202 in DraIII, positioned to coordinate the divalent metal cofactor, corresponds to N113 in PacI, N165 in Hpy99I and Q175 in KpnI, among known HNH REases (Fig. 4). H183 in DraIII corresponds to H149 in Hpy99I and H149 in KpnI, the general base that activates a nucleophile for the hydrolysis of the phosphodiester bond (Fig. 4). To verify their role in catalysis, H183 and N202 were mutated to Ala. Mutants H183A and N202A were overexpressed and were found to be inactive at the cell lysate level (Table 2). Superposition of the DraIII closed structure to Hpy99I, PacI and T4 Endo VII shows the high structural conservation of the HNH active site (Fig. 4).

Comparison of the sequence and structures of HNH REases. (A) Sequence alignment of the DraIII, Hpy99I, PacI, T4 Endo VII and KpnI HNH catalytic motif. (B–D) DraIII HNH motif (orange) aligned to Hpy991 (B: cyan, PDB 3GOX), PacI (C: green, PDB 3M7K) and T4 Endo VII (D: blue, PDB 2QNF), respectively. The zinc ion is shown as green sphere, magnesium ion and DNA are shown in red
A number of HNH endonucleases display a series of histidine and cysteine residues with a characteristic spacing that allows for coordination of a zinc ion in a cross-brace structure. Most HNH endonucleases (REases, homing endonucleases and non-specific endonucleases) contain a CXXC/CXXC zinc-binding domain in the vicinity of the HNH active site that ligates the latter to the rest of the protein. Hpy99I and PacI have an extra copy of the CXXC/CXXC domain near the N-terminus (Shen et al., 2010; Sokolowska et al., 2009). The N-terminal zinc binding site (consisting of C4, C22, H28 and C32) and the C-terminal zinc binding site (C157, C160, H198 and C201) of DraIII (Fig. 3C) deviate from the CXXC/CXXC configuration. Similar to other HNH REases and homing endonucleases, the two zinc ions are tetrahedrally coordinated by four cysteine/histidine residues in the DraIII structure (Fig. 3A).
The coordination of the zinc ion in the second zinc binding site has been shown to play an essential role in the catalytic activity of T4 Endonuclease VII (Giraud-Panis et al., 1995). The removal of the two zinc ions by denaturing the enzyme in urea has also been shown to adversely affect the folding, thermostability and cleavage activity of KpnI (Saravanan et al., 2007a). Alanine mutation of any one of the eight zinc coordinating cysteine or histidine residues rendered the DraIII enzyme inactive at the cell lysate level (Table 2).
Interactions between the N- and C-terminal domains
The DraIII structure is composed of two domains: the N-terminal domain (α1–9) and the C-terminal domain (α10–13, β1–2) (Figs. 3C and 5A). The two domains are connected by the turn between α9 and α10, opening up to the solvent at the end of α3 of the N-terminal domain and the turn connecting the two β strands of the ββα-metal fold in C-terminal domain (Fig. 5A, cartoon mode). More importantly, the domain interface opens up to the active site tunnel the substrate DNA would bind to (Fig. 5A, surface mode). Six potential hydrogen bonds are found between the two domains along the domain interface, which can be divided into three regions: the mouth, middle and hinge region (Fig. 5A). A hydrophobic patch is found at the hinge region (Fig. 5A).

The interactions between the N-terminal domain and C-terminal domain ofDraIII subunit. (A) DraIII subunit structure. N-terminal domain in orange; C-terminal domain in cyan; Zinc in green; Magnesium in red. Left: DraIII subunit displayed in surface mode. Right: DraIII subunit displayed in cartoon mode. Interactions in the mouth, middle region and hydrophobic residues in the hinge region were shown in gray boxes. The locations of potential hydrogen bonds are indicated with red dotted line. (B) Determination FI of DraIII T181A. λ DNA was cleaved by diluted T181A. Lane 1: 3.2 μmol/L T181A; Lane 13: 0.78 nmol/L (LCC); Lane 21: 3.05 pmol/L; Lane 22: 1-kb DNA Ladder. FI is larger than 4000. The hash represents a partially digested band. No star band was observed. Disrupting hydrogen bond between T181 and D55 in the middle region remarkably enhanced FI
To study the role of the potential hydrogen bonds that connect the two domains, a series of mutations were made to the hydrogen bond donors and acceptors along the domain interface. T181 was mutated to Ala to disrupt the two potential hydrogen bonds located at the middle region between T181 and D55 (Fig. 5A, middle region). The mutant T181A showed remarkable high fidelity—no star activity was observed at very high enzyme concentrations (FI ≥ 4000) (Fig. 5B). To verify the role of these hydrogen bonds in cleavage fidelity, mutations T181S and T181Y, which potentially retain the hydrogen bonds, were created. Both mutants T181Y and T181S had the same FI as the WT (Table 2). Mutating T181 further to residues that do not support hydrogen bond formation, namely, Gly, Cys, Val, Met, Leu, Asn or Gln, and D55 to Ala improved the cleavage fidelity to different extents (Table 2). Mutation of T181 to Tyr did not improve the fidelity when tested in cell lysate preparations. Unfortunately, the mutant protein T181Y could not be purified to high enough quantity for further characterization.
Mutation H189A, which potentially disrupts two potential hydrogen bonds in the middle region between H189, D55 and A56 also resulted in high fidelity (Fig. 5A and Table 2). The two potential hydrogen bonds between R187 and E107 are located at the mouth region (Fig. 5A). Mutating R187 to Ala also improved cleavage fidelity (FI ≥ 2000) (Table 2). Mutation of L129, L164, F169, or V179 in the hydrophobic patch at the hinge region to Ala also increased cleavage fidelity, albeit to a lesser extent (Table 2).
Differential scanning calorimetry (DSC) analysis of the DraIII high fidelity mutants
To evaluate the effect of the domain interface mutations on the DraIII protein, the melting temperatures (Tm) of purified DraIII WT, T181A, T181G, H189A and R187A proteins were measured by DSC. As shown in Table 3, T181A, T181G and H189A have Tm values of 56.7°C, 52.5°C and 56.2°C, respectively, compared to the WT value of 59.1°C. Mutation of T181 and H198 to Ala led to similar and lower Tm values supporting the notion that hydrogen bonds were disrupted by each of the mutations. The even lower Tm value of the mutant T181G is probably due to the thermodynamic penalty in the folding of the protein caused by the cavity created by the Gly mutation. The correlation between high cleavage fidelity and disruption of hydrogen bonds between the two domains leads us to propose that the high cleavage fidelity is the result of increased conformational flexibility of the mutant proteins. The Tm of another high fidelity mutant R187A (Tm = 60.7°C) is comparable to that of WT, suggesting that the mutation R187A improves cleavage fidelity through a different mechanism.
DISCUSSION
DraIII is the first HNH REase crystallized which recognizes a gapped sequence
The crystal structures presented here show that DraIII contains the ββα-metal fold of the HNH active site. The ββα-metal fold of DraIII is highly similar to that of known HNH enzymes such as PacI (Shen et al., 2010) and Hpy99I (Sokolowska et al., 2009) (Fig. 4). Structural superposition and sequence alignment show that N202 and H183 of DraIII correspond to the magnesium binding residue and the general base, respectively, in PacI, Hpy99I and KpnI (Fig. 4). The loss of catalytic activity when these residues were mutated is consistent with this assignment (Table 2). The HNH catalytic motif has been identified in numerous classes of nucleases, from the sequence non-specific DNA colicins (Ko et al., 1999; Pommer et al., 2001), Holliday junction-specific T4 endonuclease VII (Giraud-Panis et al., 1995), to homing endonucleases and REases (Orlowski and Bujnicki, 2008) and most recently in Cas9 of the CRISPR immunity system of bacteria and archaea (Gasiunas et al., 2012; Jinek et al., 2012). Even within the REase family the HNH enzymes exhibit diverse sequence specificity: TTA↑AT↓TAA for PacI, ↑CGWCG↓ for Hpy99I, G↑GTAC↓C for KpnI and CAC↑NNN↓GTG for DraIII. Among the known HNH REases, the structure presented here is the first for one that recognizes a gapped sequence. The absence of the DNA in the enzyme-DNA co-crystal structure, however, prevented us from examining the protein-DNA interactions through which DraIII recognizes the canonical 9-bp sequence with a three base gap in the middle.
DraIII exhibits high star activity with certain sequence specificity
Under standard reaction conditions, DraIII has a FI of 2 on λ DNA (Fig. 1A). Our investigation on the DraIII star activity revealed a few intriguing properties. First, CATNNNGTG was identified as a strong star site for DraIII. Within the context of CATNNNGTG, the DraIII star activity has a sequence preference for the central three nucleotides, whereas the canonical cleavage activity does not (Grosskopf et al., 1985). In addition, DraIII cleaves the star site in an asymmetrical manner: the canonical half site (GTG) was cleaved faster than the star half site (CAT) (Fig. 2C and 2D), similar to what had been reported with EcoRI (Lesser et al., 1990). A substrate containing pseudo-palindromic star half sites CATGTTATG was not cleaved under the same star conditions, showing that at least one copy of the canonical half site is required for double-strand cleavage. It is consistent with the absence of cleavage by EcoRI on non-canonical sites containing two non-palindromic base changes (Lesser et al., 1990).
Based on this research and previous studies on cleavage activity on star sites and crystal structures of star-substrate bound REases, a hypothesis is prompted for DraIII where a DraIII dimer scans through a piece of dsDNA through the 1-D sliding mechanism (Dikic et al., 2012; Jack et al., 1982; Rau and Sidorova, 2010; Wright et al., 1999) and pauses when a canonical half site is recognized by one of the monomers. The presence of a one-base-off half star site and a canonical half site at the correct position triggers the formation of a cleavage competent enzyme-DNA complex where each of the DraIII monomer cleaves the bound half site. The star half site is, however, cleaved at a slower rate than the canonical half site due to the thermodynamic penalty to establish non-canonical protein-DNA interactions in an “adaptive conformation” (Lesser et al., 1990; Viadiu and Aggarwal, 2000). As both strands are cleaved the target sequence is destroyed and the enzyme dissociates from the DNA. From this hypothesis, star activity of homodimeric REases is dependent on the propensity of a particular one-base-off star site to form a cleavage competent conformation with the enzyme. The thermodynamic requirements for the formation of such a conformation therefore allows some star sites to be cleaved more effectively than others. According to this hypothesis, REases showing high star activity, such as DraIII, may have a lower thermodynamic barrier to adopt the cleavage competent conformation with some star sites. Our results suggest that the sequence of the middle three nucleotides in the context of a star site also contributes to the energy barrier in forming this conformation.
Eliminating inter-domain interaction improves the fidelity of DraIII restriction endonuclease
According to our hypothesis of how star sites are cleaved, one can prevent cleavage at star sites by creating a higher thermodynamic barrier for the enzyme and the bound star substrate to achieve the cleavage competent state, such that favorable interactions between the enzyme and the cognate substrate can generate enough binding energy to overcome the higher thermodynamic barrier, whereas the lack of such interactions between the enzyme and star substrate results in abolition of cleavage.
In this work it was shown that cleavage fidelity of DraIII can be improved by mutation of residues involved in the interactions between the two structural domains (Table 2 and Fig. 5). The potential hydrogen bonds between T181 and D55 in the middle region was studied in most detail. Either removing the hydrogen donor or acceptor (mutations T181A, T181G, T181V, T181M, T181C, T181L and D55A) remarkably enhanced cleavage fidelity. Thus, we hypothesize that these interactions, which also make the protein structure less flexible, help to lower the thermodynamic barrier of the DraIII-DNA complex to achieve the cleavage competent state. The low thermodynamic barrier allows the DraIII-star substrate complex to reach the cleavage competent state with less favorable binding interactions, hence the relatively high star activity of DraIII. Disruption of these interactions in the high fidelity mutants on the other hand imposes a higher thermodynamic barrier such that only the binding of the canonical substrate but not the star substrates can generate enough favorable energy to reach the cleavage competent state. This hypothesis is consistent with the higher free energy changes required for the formation of the cleavage-competent transition state for EcoRI with star sites than with the canonical site (Lesser et al., 1990), and could potentially be applicable to the star activity of other type IIP REases in general.
It should be pointed out that mutations that reduce the hydrophobicity and steric integrity of the hydrophobic patch located at the hinge region of the interface (L129A, L164A, F169A and V179A) also improved DraIII fidelity albeit to lesser extent (Table 2). On the other hand, mutant R187A has a Tm value similar to the wild type value (Table 3), suggesting that the hypothesized hydrogen bonds between R187 and E107 are neither present nor important for folding in native DraIII. From the DraIII structure derived from the protein-DNA co-crystal, R187 potentially interacts with the substrate DNA (Fig. 5A, mouth region). Hence, mutation R187A improves fidelity probably through altering the protein-DNA interaction network. The mechanism of this fidelity improvement will be studied elsewhere.
In this paper, we reported that eliminating inter-domain interaction improves the fidelity of DraIII restriction endonuclease. The understanding of fidelity mechanism could be applicable to the star activity of other REases and help to engineer high fidelity REases.
MATERIALS AND METHODS
Strain construction, protein expression and purification
The DraIII endonuclease gene draIIIR was inserted into pAGR3 vector to create the plasmid pAGR3-draIIIR. The DraIII methyltransferase gene draIIIM was inserted into pACYC184 to create the plasmid pACYC184-draIIIM. E. coli C3081 (NEB) was first transformed by pACYC184-draIIIM so that the DraIII sites in the host genome could be modified by the constitutively expressed DraIII methyltransferase controlled by the Tet promoter. The transformed C3081 was further transformed by pAGR3-draIIIR to create a stable DraIII endonuclease expressing strain. The strain was grown in LB containing ampicillin and chloramphenicol at 37°C. Protein expression was induced by adding IPTG to 0.5 mmol/L and further incubation for 16 h at 37°C. The collected cells were suspended in Buffer A (20 mmol/L potassium phosphate, 100 mmol/L NaCl, 0.1 mmol/L EDTA, 10 mmol/L 2-mercaptoethanol, 5% glycerol, pH7.0) containing 1 mmol/L PMSF before the cells were disrupted by sonication. After centrifugation the supernatant was loaded onto a Heparin HyperD column (Sigma) and eluted with a linear gradient of Buffer B (20 mmol/L potassium phosphate, 1 mol/L NaCl, 0.1 mmol/L EDTA, 10 mmol/L 2-mercaptoethanol, 5% glycerol, pH7.0). Active fractions were eluted at approximately 500 mmol/L NaCl. These fractions were pooled, diluted 5-fold and loaded onto a Source 15S column (GE Healthcare). Protein was eluted with the same NaCl gradient. Active fractions eluted at approximately 300 mmol/L NaCl. Proteins were concentrated and stored. Mutations were constructed by inverse PCR using pAGR3-draIIIR as template. The DraIII mutants were expressed and purified using the same methods as for wild type (WT).
Fidelity Index (FI) determination
To measure the FI of DraIII and its mutants, a two-fold dilution series of the concentrated protein stock solution was made using Diluent B (300 mmol/L NaCl, 10 mmol/L Tris-Cl, 0.1 mmol/L EDTA, 1 mmol/L dithiothreitol, 500 μg/mL BSA, 50% glycerol; NEB) to give 21 decreasing concentrations of the enzyme (1×, 0.5×, 0.25×, etc.). Two microliters of the diluted enzyme solutions were then mixed with 1 μg λ DNA in a reaction volume of 20 μL in NEBuffer 4 (50 mmol/L potassium acetate, 20 mmol/L Tris-acetate, 10 mmol/L magnesium acetate, 1 mmol/L dithiothreitol, pH 7.9 at 25°C; NEB). The reactions were incubated at 37°C for 1 h and were then quenched by 2 μL of Stop Solution (10×, 200 mmol/L EDTA, 100 mmol/L Tris-Cl, pH 8.0, 0.03% bromophenol blue, 0.94% SDS). The quenched reactions were analyzed by agarose gel electrophoresis. Gel images were obtained using a UV imager (Bio-Rad) on ethidium bromide (EB)-stained gels. The FI was calculated as the ratio of the highest enzyme dilution showing no star activity to the lowest dilution showing complete digestion (Wei et al., 2008). The FIs were determined with purified proteins except for T181Y. The FI of T181Y was determined using cell lysate dilutions.
DNA cleavage assays
The DraIII star sites on pUC19 and pXba were located by incubating the respective plasmid DNAs (1 μg) in 20 μL reactions containing BamHI (20 units), XmnI (20 units) and/or DraIII (2 pmol) and 2 μL of 10× NEBuffer 4 at 37°C for 1 h. DNA was analyzed by 0.8% agarose gel electrophoresis followed by EB staining and UV imaging.
The 5′-Cy5 labeled 39-bp DNA duplexes and 15, 16, 17, 25, 26, 27-nt markers were synthesized by Sangon. 100 nmol/L of the canonical or star DNA was incubated with 500 nmol/L of DraIII WT in NEBuffer 4 at 37°C for 1 h. The reactions were stopped by adding EDTA to 20 mmol/L. Samples were then analyzed by 20% TBE urea polyacrylamide gel electrophoresis. The 5′-Cy5 labeled 39-bp DNA duplexes were used for time-course experiments. 100 nmol/L of star DNA and 500 nmol/L DraIII WT (or 100 nmol/L of canonical DNA and 10 nmol/L DraIII WT) were incubated in NEBuffer 4 at 37°C. Samples collected at the designated time intervals were stopped by adding EDTA to 20 mmol/L. The reactions were then analyzed by 15% TBE urea polyacrylamide gel electrophoresis (Bio-Rad). Gel images were obtained using the Typhoon TRIO Variable Mode Imager (GE Healthcare) using 633 nm excitation. The quantification of products was analyzed using the ImageQuant TL software (GE Healthcare).
The 29-bp DNA duplexes were digested by DraIII in 20 μL reactions containing 5 μmol/L DraIII, 2.5 μmol/L DNA and 2 μL 10× NEBuffer 4 at 37°C for 2 h. The reactions were stopped by adding 2 μL of Stop Solution and analyzed by electrophoresis on 10% non-denaturing TBE polyacrylamide gel. The gels were stained by EB and images obtained by UV imaging.
Comparison of endonuclease activity of DraIII proteins at cell lysate level
DraIII mutants were overproduced using the same conditions as the WT enzyme. One milliliter of culture cells was collected and suspended in 150 μL Buffer A and 50 μL glass beads (Sigma). Cells were broken by vortex and cell lysate was collected after centrifugation. The cell lysate was then diluted 1, 10, 102 and 103 folds using Diluent B. Two microliters of the diluted cell lysate solutions were then mixed with 1 μg λ DNA in 20 μL reactions in NEBuffer 4. The reactions were incubated at 37°C for 1 h and then quenched by Stop Solution. The reactions were analyzed by agarose gel electrophoresis.
Crystallization
DraIII and the 11-bp phosphothioate canonical DNA duplex at a 1:1 molar ratio were incubated on ice for one hour and then subjected to size exclusion chromatography on a HiLoad superdex 75 gel filtration column (GE Healthcare) in the presence of 20 mmol/L Tris-Cl, 150 mmol/L NaCl, 5 mmol/L MgCl2, 5 mmol/L DTT, pH 7.0. The target DraIII and DNA complex was concentrated to 12 mg/mL for crystallization. Crystals were grown at 18°C by the hanging-drop vapor-diffusion method in a reservior solution containing 0.1 mol/L Tris-Cl, 20% 2-propanol, 5% PEG 8000, pH8.0, and then were cryo-protected using a reservoir solution supplemented with 25% (v/v) glycerol and then flash-frozen in liquid nitrogen. The crystals belonged to space group P2(1).
Structure determination
All the data were collected at the Shanghai Synchrotron Radiation Facility (SSRF) BL17U, and integrated and scaled using the HKL2000 package (Otwinowski and Minor, 1997). Further processing was carried out using programs from the CCP4 suite (Collaborative Computational Project, 1994). Data collection statistics are summarized in Table 1. The real-space constraints were applied to the electron density map in DM (Cowtan, 1994). The final electron density map was of sufficient quality for BUCCANEER (Cowtan, 2006) to be able to build almost the complete model. The final model rebuilding was performed using COOT (Emsley and Cowtan, 2004) and the protein structure was refined with PHENIX (Adams et al., 2002) using NCS and stereochemistry information as restraints. Structural figures were generated in PyMOL (http://www.pymol.org).
Differential scanning calorimetry (DSC)
Purified proteins were dialyzed into DSC buffer (20 mmol/L potassium phosphate, 200 mmol/L NaCl, 10 mmol/L MgCl2, 5% glycerol, pH 7.0) and protein concentration is adjusted to about 6 mg/mL. DSC experiments were performed using a MicroCal VP-Capillary DSC system (GE Healthcare). The experimental scan rate was set at 90°C/h with a temperature range from 10°C to 110°C. The DSC data were analyzed using the Origin 7.1 software and fitted using a two-state model. The Origin analysis reported values for the Tm (melting temperature) for each observed transition.
PROTEIN DATA BANK ACCESSION NUMBERS
The DraIII protein structure has been deposited in the Protein Data Bank (http://www.rcsb.org/) with accession number 4L0K.
Abbreviations
- DSC:
-
differential scanning calorimetry
- FI:
-
Fidelity Index
- REases:
-
restriction endonucleases
- Tm:
-
melting temperature
References
Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr 58:1948–1954
Chan SH, Opitz L, Higgins L, O’Loane D, Xu SY (2010) Cofactor requirement of HpyAV restriction endonuclease. PLoS ONE 5:e9071
Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr 50:760–763
Cowtan KD (1994) DM: an automated procedure for phase improvement by density modification. CCP4 Newslett 31:34–38
Cowtan K (2006) The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr 62:1002–1011
Dikic J, Menges C, Clarke S, Kokkinidis M, Pingoud A, Wende W, Desbiolles P (2012) The rotation-coupled sliding of EcoRV. Nucleic Acids Res 40:4064–4070
Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr 60:2126–2132
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci USA 109:E2579–E2586
George J, Chirikjian JG (1982) Sequence-specific endonuclease BamHI: relaxation of sequence recognition. Proc Natl Acad Sci USA 79:2432–2436
Giraud-Panis MJ, Duckett DR, Lilley DM (1995) The modular character of a DNA junction-resolving enzyme: a zinc-binding motif in bacteriophage T4 endonuclease VII. J Mol Biol 252:596–610
Grosskopf R, Wolf W, Kessler C (1985) Two new restriction endonucleases Drall and DrallI from Deinococcus radiophilus. Nucleic Acids Res 13:1517–1528
Halford SE, Lovelady BM, McCallum SA (1986) Relaxed specificity of the EcoRV restriction endonuclease. Gene 41:173–181
Horton JR, Blumenthal RM, Cheng X (2004) Restriction endonucleases: structure of the conserved catalytic core and the role of metal ions in DNA cleavage. In: Pingoud AM (ed) Restriction endonucleases. Springer, Berlin, pp 361–392
Ibryashkina EM, Zakharova MV, Baskunov VB, Bogdanova ES, Nagornykh MO, Den’mukhamedov MM, Melnik BS, Kolinski A, Gront D, Feder M et al (2007) Type II restriction endonuclease R. Eco29kI is a member of the GIY-YIG nuclease superfamily. BMC Struct Biol 7:48
Jack WE, Terry BJ, Modrich P (1982) Involvement of outside DNA sequences in the major kinetic path by which EcoRI endonuclease locates and leaves its recognition sequence. Proc Natl Acad Sci USA 79:4010–4014
Jakubauskas A, Giedriene J, Bujnicki JM, Janulaitis A (2007) Identification of a single HNH active site in type IIS restriction endonuclease Eco31I. J Mol Biol 370:157–169
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Kaminska KH, Kawai M, Boniecki M, Kobayashi I, Bujnicki JM (2008) Type II restriction endonuclease R. Hpy188I belongs to the GIY-YIG nuclease superfamily, but exhibits an unusual active site. BMC Struct Biol 8:48
Ko TP, Liao CC, Ku WY, Chak KF, Yuan HS (1999) The crystal structure of the DNase domain of colicin E7 in complex with its inhibitor Im7 protein. Structure 7:91–102
Kriukiene E (2006) Domain organization and metal ion requirement of the Type IIS restriction endonuclease MnlI. FEBS Lett 580:6115–6122
Lesser DR, Kurpiewski MR, Jen-Jacobson L (1990) The energetic basis of specificity in the EcoRI endonuclease–DNA interaction. Science 250:776–786
Malyguine E, Vannier P, Yot P (1980) Alteration of the specificity of restriction endonucleases in the presence of organic solvents. Gene 8:163–177
Mehta P, Katta K, Krishnaswamy S (2004) HNH family subclassification leads to identification of commonality in the His-Me endonuclease superfamily. Protein Sci 13:295–300
Miyazono K, Watanabe M, Kosinski J, Ishikawa K, Kamo M, Sawasaki T, Nagata K, Bujnicki JM, Endo Y, Tanokura M et al (2007) Novel protein fold discovered in the PabI family of restriction enzymes. Nucleic Acids Res 35:1908–1918
Nasri M, Thomas D (1987) Alteration of the specificity of PvuII restriction endonuclease. Nucleic Acids Res 15:7677–7687
Orlowski J, Bujnicki JM (2008) Structural and evolutionary classification of Type II restriction enzymes based on theoretical and experimental analyses. Nucleic Acids Res 36:3552–3569
Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276:307–326
Pommer AJ, Cal S, Keeble AH, Walker D, Evans SJ, Kuhlmann UC, Cooper A, Connolly BA, Hemmings AM, Moore GR et al (2001) Mechanism and cleavage specificity of the H-N-H endonuclease colicin E9. J Mol Biol 314:735–749
Rau DC, Sidorova NY (2010) Diffusion of the restriction nuclease EcoRI along DNA. J Mol Biol 395:408–416
Roberts RJ (2005) How restriction enzymes became the workhorses of molecular biology. Proc Natl Acad Sci USA 102:5905–5908
Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev S, Dryden DT, Dybvig K et al (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31:1805–1812
Robinson CR, Sligar SG (1995) Heterogeneity in molecular recognition by restriction endonucleases: osmotic and hydrostatic pressure effects on BamHI, PvuII, and EcoRV specificity. Proc Natl Acad Sci USA 92:3444–3448
Sapranauskas R, Sasnauskas G, Lagunavicius A, Vilkaitis G, Lubys A, Siksnys V (2000) Novel subtype of type IIs restriction enzymes. BfiI endonuclease exhibits similarities to the EDTA-resistant nuclease Nuc of Salmonella typhimurium. J Biol Chem 275:30878–30885
Saravanan M, Bujnicki JM, Cymerman IA, Rao DN, Nagaraja V (2004) Type II restriction endonuclease R. KpnI is a member of the HNH nuclease superfamily. Nucleic Acids Res 32:6129–6135
Saravanan M, Vasu K, Ghosh S, Nagaraja V (2007a) Dual role for Zn2+ in maintaining structural integrity and inducing DNA sequence specificity in a promiscuous endonuclease. J Biol Chem 282:32320–32326
Saravanan M, Vasu K, Kanakaraj R, Rao DN, Nagaraja V (2007b) R. KpnI, an HNH superfamily REase, exhibits differential discrimination at non-canonical sequences in the presence of Ca2+ and Mg2+. Nucleic Acids Res 35:2777–2786
Shen BW, Heiter DF, Chan SH, Wang H, Xu SY, Morgan RD, Wilson GG, Stoddard BL (2010) Unusual target site disruption by the rare-cutting HNH restriction endonuclease PacI. Structure 18:734–743
Sokolowska M, Czapinska H, Bochtler M (2009) Crystal structure of the beta beta alpha-Me type II restriction endonuclease Hpy99I with target DNA. Nucleic Acids Res 37:3799–3810
Viadiu H, Aggarwal AK (2000) Structure of BamHI bound to nonspecific DNA: a model for DNA sliding. Mol Cell 5:889–895
Vincze T, Posfai J, Roberts RJ (2003) NEBcutter: a program to cleave DNA with restriction enzymes. Nucleic Acids Res 31:3688–3691
Wei H, Therrien C, Blanchard A, Guan S, Zhu Z (2008) The fidelity index provides a systematic quantitation of star activity of DNA restriction endonucleases. Nucleic Acids Res 36:e50
Wright DJ, Jack WE, Modrich P (1999) The kinetic mechanism of EcoRI endonuclease. J Biol Chem 274:31896–31902
Acknowledgements
D.S. and X.L. designed research; X.L. and W.Z. performed research; L.Z., F.L., Z.Z. and M.Y. contributed new reagents/analytic tools; M.Y., X.L., D.S. and S.C. wrote the paper.
Work carried out at New England Biolabs Shanghai R&D Center was supported by New England Biolabs, Inc. Work carried out at the Yang lab in Tsinghua University was supported by the National Basic Research Program (973 Program) (Nos. 2011CB910502 and 2012CB911101) and the National Natural Science Foundation of China (Grant Nos. 31030020 and 31170679). The authors thank the staff at the SSRF BL17U beamline for their assistance in data collection.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Wei Zhuo and Xuhui Lai have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Zhuo, W., Lai, X., Zhang, L. et al. Elimination of inter-domain interactions increases the cleavage fidelity of the restriction endonuclease DraIII. Protein Cell 5, 357–368 (2014). https://doi.org/10.1007/s13238-014-0038-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-014-0038-z