
Abstract
Extramedullary hematopoiesis (EMH), as a compensatory phenomenon, refers to the blood cell formation outside of the bone marrow that occurs once the cells in the circulatory system fail to meet individuals’ needs. EMH is rare in moderate to severe beta thalassemia because most symptomatic patients are effectively managed with transfusion. However, patients that fail to receive transfusions like β-thalassemia intermedia (β-TI) as indicated are at increased risk for developing EMH. This paper describes the case of a 15-year-old female adolescent with β-thalassemia major (β-TM), suffering from a rare form of EMH affecting the sinus cavities, characterized by headache, sinusitis, and nasal obstruction, as confirmed by physical-pathological examinations and computerized tomography (CT) scan findings. The EMH in this patient could be significantly attributed to the lack of regular blood transfusions in recent years. It was concluded that β-TM along with the occurrence of EMH in the sinus cavities had led to a complex case, carrying a heavy burden of the disease for the patient.
Similar content being viewed by others
Introduction
Hematopoiesis is the process of blood cell formation that begins in the yolk sac in the embryonal period, followed by production in the liver, and ultimately occurring almost exclusively in the bone marrow past the age of five. In spite of this, some conditions, such as inflammation, anemia (e.g., due to thalassemia), myelofibrosis, and certain pathological factors may induce hematopoiesis outside of the bone marrow. This condition, called extramedullary hematopoiesis (EMH), was first illustrated by Guizetti in 1912 [1, 2]. EMH represents a nonneoplastic compensatory mechanism that arises once the bone marrow fails to produce enough blood cells [3]. While EMH can have effect in different parts of the body, it is much more common in certain organs, like the liver and spleen [4]. EMH also tends to be latent and asymptomatic but problematic for some patients. For example, there are several case reports of severe neurological symptoms due to EMH in the spinal cord [5, 6]. There is even one case report of EMH in the brain of a patient with β-T, causing ossicular fixation and bilateral conductive hearing loss [7]. In one patient with β-TI, thoracic EMH had thus led to shortness of breath and dry cough [8]. Furthermore, EMH occurring due to the formation of tumefactive masses may make it more difficult to perform a differential diagnosis among pathologies and tumors with similar conditions, which highlights the importance of paying much attention to EMH. β-TM is further a serious disease that requires the affected patients to undergo regular blood transfusions; the risk of developing EMH in these patients is very low. Because the body’s deficiency of need is compensated by blood transfusions. This paper describes the case of a patient with β-TM who has developed EMH in her sinus cavities (viz. maxillary, ethmoid, and sphenoid) most likely because of not undergoing regular blood transfusions in recent years.
Case presentation
The patient here was a 15-year-old Iranian female adolescent suffering from β-TM (IVSII-I(G-A)/IVSII(G-A)- β0/β0) admitted for facial swelling and splenomegaly to Shafa Hospital, affiliated to Jundishapur University of Medical Sciences, Ahvaz, Iran.
Despite needing regular blood transfusions, the patient had not undergone any blood transfusions in recent years. Moreover, examinations revealed pallor and jaundice, bilateral maxillary swelling, and thalassemic facies (Fig. 1). Of note, the patient was 145 cm tall and weighed 35 kg, which was not proportional to her age.

Bilateral maxillary swelling, swollen gums, and thalassemic facies
The ultrasound results also showed splenomegaly of size 58 × 129 mm and hepatomegaly of size 132 mm. As well, the brain magnetic resonance imaging (MRI) revealed the asymmetric swelling of the sinuses, putting more pressure on the nasal septum and causing a deviated septum.
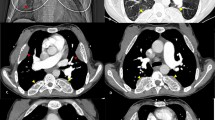
The maxillary sinuses of both sides had been dilated because of hyperplasia and had a reduced wall thickness (Fig. 2). Hyperplasia was even more intense in the right maxillary sinus, leading to the closure of the right airway. Although this condition was also present in the left-side maxillary sinus, it was mild. The dilatation of the right-side maxillary sinus had further put pressure on the nasal septum, causing nasal deviation. Moreover, the calvaria had an irregular texture with the signs of thickening and a spongy appearance in the frontal and posterolateral sections. Accordingly, it was concluded that the progression of this condition could bring about head deformity.

CT scan showed dilation, thinning, and opacification in the right ethmoid sinus (yellow arrow). The sphenoid sinus was also fully opacified, with a spongy appearance (red arrow)
The laboratory findings were as follows:
Blood urea nitrogen (BUN), 15 mmol/L; creatinine (CR), 1.0 mg/dL; bilirubin (BR, total), 4.1 µmol/L; BR (direct), 0.4 mg/dL; aspartate transaminase (AST), 40 U/L; alanine aminotransferase (ALT), 38 IU/L; alkaline phosphatase (ALP), 535 IU/L; phosphor (P), 5.4 mg/dL; ferritin (FER), 311.1 ng/mL; 25-OH-vitamin D, 17.4 ng/mL; white blood count (WBC), 8.500 × 109/L; red blood count (RBC), 3.81 × 1012/L; hemoglobin (Hb), 9.2 g/dL; mean corpuscular volume (MCV) 72.4 fl; mean corpuscular hemoglobin (MCH), 24.1 pg; mean corpuscular hemoglobin concentration (MCHC), 33.3 g/dL; red cell distribution width-coefficient of distribution (RDW-CV), 28.9%; RDW-standard deviation (RDW-SD), 63.8 fL; and platelet (PLT) 318 × 109/L. In addition, the electrophoresis outcomes revealed fetal hemoglobin (HbF), 98.1%, and hemoglobin A2 (HbA2), 1.9% (Fig. 3).

The graph for the patient’s electrophoresis results
For the final diagnosis, a biopsy was taken from the patient’s sinuses that it showed the presence of trilineage hematopoiesis that confirmed the diagnosis of EMH (Fig. 4). The patient was also prescribed folic acid 5 mg/day, hydroxyurea 500 mg/day, and zinc-plus, and then received 10 sessions of 10 Gray (GY) radiotherapy. After 9 months, the size of the mass has slightly decreased, and the patient undergoes surgery in the next stage.

The specimen received in the fixative formalin, consisting of multiple tiny fragments of the bone and the soft tissue totally M: 1 × 1 × 0.5 cm. The sections showed sinonasal mucosa and connective tissues with mild inflammation, admixed with the fragments of the bony trabeculae with areas of trilineage hematopoiesis, including erythroid, myeloid, and megakaryocytic, which confirmed the diagnosis of EMH
Discussion and conclusion
A small amount of hematopoietic stem cells (HSCs) circulates in the peripheral blood and the spleen in a steady state. Furthermore, in patients with TM, who stop undergoing blood transfusions, the bone marrow ramps up to overcome chronic anemia, which leads to osteoporosis and bone deformities [3]. This causes erythroid precursors to separate from the bone marrow and move to other organs. The liver and spleen are also two organs with the highest likelihood of EMH because of their filtration capability, which causes the entrapment and homing of erythroid precursors in these tissues, ultimately inducing hepatosplenomegaly [3]. However, this does not mean that EMH cannot take place in other organs, and indeed this condition can occur, as was the case for the patient here, because of the formation of erythroid mass and the start of hematopoiesis in a tertiary site (e.g., the sinus cavity) in response to the inability to meet circulatory demands.
Recently emerged molecular evidence has further shed some light on the molecular background of EMH. Some studies have pointed to several genetic factors involved in the EMH progression, including Ras homolog enriched in brain 1 (Rheb1) loss, ASXL transcriptional regulator 2 (ASXL2) deficiency, zinc finger E-box binding homeobox 2 (Zeb2) inactivation, and SET domain containing 1B, histone lysine methyltransferase (Setd1b) deletion [9]. These molecules appear to be part of the network that regulates hematopoiesis. For example, Setd1b has a regulatory effect on key lineage specification components, such as CCAAT enhancer-binding protein alpha (Cebpa), GATA-binding protein 1 (Gata1), and Kruppel-like factor 1 (klf1), and results in the loss of expression in myeloid-biased mice EMH in the spleen [10].
In one study, a correlation had been accordingly found between the T-cell leukemia homeobox 1 (Tlx1) expression in mesenchymal progenitor-like cells in the spleen along with the recruitment and mobilization of circulating hematopoietic stem/progenitor cells (HSPCs) in this organ, which seemed to be involved in EMH in the spleen [9]. In fact, it appears that niche-forming cells, such as mesenchymal stem/progenitor cells (MSPCs) located in places other than the bone marrow play a significant role in the recruitment and cell fate processes as well as the triggering of EMH in the organ [1].
It has been also reported that nitrogen-containing bisphosphonate (NBP) medications, such as alendronate, neridronate, and ibandronate, often used to treat osteoporosis in thalassemia patients can augment the risk of EMH by inducing histidine decarboxylase (HDC) production [11,12,13].
Histidine decarboxylase (HDC) is a primary and unique enzyme that catalyzes the synthesis of histamine through the decarboxylation of the amino acid L-histidine in mouse and human.
HDC is expressed in the hematopoietic progenitor cell. Comparison of morphological analysis findings in the liver of HDC-knockout mice with wild-type showed more significant hematopoietic colonies and megakaryocytes in the former. It seems that high levels of HDC in the culture medium cause detachment of hematopoietic stem cells and their disappearance [14]. On the other hand, HDC as wells also involved in maintaining a suitable environment for hematopoiesis by inducing the expression of interleukin 3 (IL-3), IL-17, granulocyte colony-stimulating factor (G-CSF), erythropoietin (EPO), and C-X-C motif chemokine ligand 12 (CXCL12) [14, 15]. As well, EPO is one of the main factors affecting the onset of EMH, and anemic patients show increased EPO expression due to lower oxygen delivery to the tissues in the liver and the kidneys regardless of the NBP consumption. Moreover, EPO plays a direct role in the release of HSPCs from the bone marrow and the rise of the HSPC level in circulation [16]. EPO similarly activates the erythropoietin receptor/Janus kinase 2 (EPOR/JAK2) pathway, whose signaling leads to the excessive proliferation of erythroid cells and increases the number of these cells in the secondary hematopoietic organ [9, 17].
While the common practice for the final diagnosis of EMH is a biopsy, other noninvasive methods such as CT scan, MRI, and chest roentgenograms can be effective [5, 8]. One of the major challenges to deal with EMH is the difficulty of early detection and diagnosis. In a study comparing the levels of different factors in non-transfusion-dependent thalassemia (NTDT) patients, EMH-positive cases had thus shown significantly higher serum FER, growth differentiation factor-15 (GDF-15), and EPO levels than EMH-negative ones [18]. This study had also reported that GDF15, EPO, GDF15/EPO, and GDF15/soluble transferrin receptors (sTfR) could be exploited for the early risk prediction of EMH in the NTDT patients [18]. In another study, sTfR could act as a prognostic factor for EMH in β-TI patients [19]. In view of this, these factors can be used as prognostic markers to help identify patients with a high risk of EMH in order to prevent an elevation in the burden of the disease.
In general, there are still no clear guidelines for treating EMH, and physicians often consider radiotherapy (10–30 Gy), surgical interventions, hydroxyurea therapy, hyper-transfusion, or a combination of these methods as treatment options [20]. However, some methods have certain drawbacks that make them less preferable. For example, while blood stem cells are highly sensitive to radiotherapy, this treatment can cause secondary bone marrow capacity reduction. Similarly, surgical interventions can be associated with the risk of excessive bleeding [21]. Furthermore, there is a significant risk of recurrence (19–37%) in patients undergoing surgery or radiotherapy for EMH [5, 8]. Thus, it seems that a more appropriate method for dealing with EMH is to utilize a combination therapy comprised of—for example—hydroxyurea therapy or EPO treatment with radiotherapy (22). In any case, there is a dire need for a gold standard for the treatment of EMH in order to reduce the burden of this condition. It might be also helpful to develop a diagnostic method or a score system for identifying patients with a high risk of EMH.
Data availability
The data and materials were taken from medical records and archives.
References
Oda A, Tezuka T, Ueno Y, Hosoda S, Amemiya Y, Notsu C et al (2018) Niche-induced extramedullary hematopoiesis in the spleen is regulated by the transcription factor Tlx1. Sci Rep 8(1):1–16
Kim CH (2010) Homeostatic and pathogenic extramedullary hematopoiesis. Journal of blood medicine 1:13
Malla S, Razik A, Das C, Naranje P, Kandasamy D, Kumar R (2020) Marrow outside marrow: imaging of extramedullary haematopoiesis. Clin Radiol 75(8):565–578
Tabesh H, Shekarchizadeh A, Mahzouni P, Mokhtari M, Abrishamkar S, Abbasi FS (2011) An intracranial extramedullary hematopoiesis in a 34-year-old man with beta thalassemia: a case report. J Med Case Reports 5(1):1–4
Yathiraj PH, Singh A, Vidyasagar S, Varma M, Mamidipudi V (2016) Excellent and durable response to radiotherapy in a rare case of spinal cord compression due to extra-medullary hematopoiesis in β-thalassemia intermedia: case report and clinicoradiological correlation. Annals of palliative medicine 6(2):195–199
Keikhaei B, Zandian K, Rahim F (2008) Existence of cord compression in extramedullary hematopoiesis due to beta thalassemia intermedia. Hematology 13(3):183–186
Lanigan A, Fordham MT (2017) Temporal bone extramedullary hematopoiesis as a causeof pediatric bilateral conductive hearing loss: case report and review of the literature. Int J Pediatr Otorhinolaryngol 97:135–138
Abdulla MA, Yassin MA, Abdelrazek M, Mudawi D, Ibrahim F, Soliman DS et al (2018) A persistent cough as atypical clinical presentation of intrathoracic extramedullary hematopoiesis (EMH) in a female with thalassemia intermedia. Acta Bio Medica: Atenei Parmensis 89(Suppl 2):41
Yang X, Chen D, Long H, Zhu B (2020) The mechanisms of pathological extramedullary hematopoiesis in diseases. Cell Mol Life Sci 77(14):2723–2738
Schmidt K, Zhang Q, Tasdogan A, Petzold A, Dahl A, Arneth BM et al (2018) The H3K4 methyltransferase Setd1b is essential for hematopoietic stem and progenitor cell homeostasis in mice. Elife 7:e27157
Otsuka H, Endo Y, Ohtsu H, Inoue S, Noguchi S, Nakamura M et al (2021) Histidine decarboxylase deficiency inhibits NBP-induced extramedullary hematopoiesis by modifying bone marrow and spleen microenvironments. Int J Hematol 113(3):348–361
Kowsaryan M, Zafari M (2016) Which pamidronate protocol is the best for treating osteoporosis in beta-thalassemia major? Ann Hematol 95(3):383–386
Forni GL, Perrotta S, Giusti A, Quarta G, Pitrolo L, Cappellini MD et al (2012) Neridronate improves bone mineral density and reduces back pain in β-thalassaemia patients with osteoporosis: results from a phase 2, randomized, parallel-arm, open-label study. Br J Haematol 158(2):274–282
Otsuka H, Endo Y, Ohtsu H, Inoue S, Kuraoka M, Koh M et al (2021) Changes in histidine decarboxylase expression influence extramedullary hematopoiesis in postnatal mice. Anat Rec 304(5):1136–1150
Horváth Z, Pállinger É, Horváth G, Jelinek I, Veszely G, Fűrész J et al (2010) Extramedullary hematopoiesis is dysregulated in histamine-free histidine decarboxylase knockout (HDC−/−) mice. Inflamm Res 59(6):429–436
Ricchi P, Meloni A, Grigoratos C, Toia P, Fina P, Pistoia L et al (2019) Prevalence of extramedullary hematopoiesis, renal cysts, splenic and hepatic lesions, and vertebral hemangiomas among thalassemic patients: a retrospective study from the Myocardial Iron Overload in Thalassemia (MIOT) network. Ann Hematol 98(6):1333–1339
Kessinger A, Bishop M, Jackson J, O’Kane-Murphy B, Vose J, Bierman P et al (1995) Erythropoietin for mobilization of circulating progenitor cells in patients with previously treated relapsed malignancies. Exp Hematol 23(7):609–612
Huang Y, Liu R, Wei X, Liu J, Pan L, Yang G, et al. Erythropoiesis and iron homeostasis in non-transfusion-dependent thalassemia patients with extramedullary hematopoiesis. BioMed research international. 2019;2019
Ricchi P, Ammirabile M, Costantini S, Di Matola T, Verna R, Diano A et al (2012) A useful relationship between the presence of extramedullary erythropoeisis and the level of the soluble form of the transferrin receptor in a large cohort of adult patients with thalassemia intermedia: a prospective study. Ann Hematol 91(6):905–909
Hisamud-Din N, Mustafah NM, Fauzi AA, Hashim NM (2017) Incomplete paraplegia caused by extramedullary hematopoiesis in a patient with thalassemia intermedia. Spinal Cord Series and Cases 3(1):1–3
Musallam KM, Taher AT, Rachmilewitz EA (2012) β-thalassemia intermedia: a clinical perspective. Cold Spring Harb Perspect Med 2(7):a013482
Goerner M, Gerull S, Schaefer E, Just M, Sure M, Hirnle P (2008) Painful spinal cord compression as a complication of extramedullary hematopoiesis associated with β-thalassemia intermedia. Strahlenther Onkol 184(4):224–226
Acknowledgements
The authors thank their colleagues in Shafa Hospital for their collaboration.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics approval
Written informed consent was obtained from the patient and her parents for the publication of this case report as well as any accompanying images. A copy of the written consent is available for review by the editor-in-chief of this journal.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Keikhaei, B., Purrahman, D., Marashi, B. et al. Extramedullary hematopoiesis in β-thalassemia major patient: a case report and review of the literature. J Hematopathol 15, 185–190 (2022). https://doi.org/10.1007/s12308-022-00506-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12308-022-00506-7