
Abstract
Currently there is no effective antiviral therapy for SARS-CoV-2 infection, which frequently leads to fatal inflammatory responses and acute lung injury. Here, we discuss the various mechanisms of SARS-CoV-mediated inflammation. We also assume that SARS-CoV-2 likely shares similar inflammatory responses. Potential therapeutic tools to reduce SARS-CoV-2-induced inflammatory responses include various methods to block FcR activation. In the absence of a proven clinical FcR blocker, the use of intravenous immunoglobulin to block FcR activation may be a viable option for the urgent treatment of pulmonary inflammation to prevent severe lung injury. Such treatment may also be combined with systemic anti-inflammatory drugs or corticosteroids. However, these strategies, as proposed here, remain to be clinically tested for effectiveness.
Similar content being viewed by others
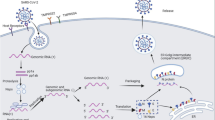

The newly emerging coronavirus, severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), causes fatal acute respiratory disease (ARD) resembling that of SARS-CoV (Chen et al.2020; Guan et al.2020; Huang et al.2020). The pathophysiology for SARS-CoV-2 has not been well studied, but likely resembles that of SARS-CoV; the acute lung injury caused by SARS-CoV infection mainly results from aggressive inflammation initiated by viral replication (Wong et al.2004). Similar to SARS-CoV infection, SARS-CoV-2 infection also causes increased secretion of IL-1β, IFN-γ, IP-10, MCP-1, IL-4, and IL-10 (Huang et al.2020). In addition, compared with non-ICU (intensive care unit) patients, ICU patients with severe disease had higher plasma levels of IL-2, IL-7, IL-10, GCSF, IP-10, MCP-1, MIP-1A, and TNF-α, suggesting a possible cytokine storm associated with disease severity (Huang et al.2020). Nevertheless, the causes of these exuberant inflammatory responses in SARS-CoV-2 infection remain largely unknown. In this review, we attempt to discuss and summarize possible mechanisms of SARS-CoV-2-mediated inflammatory responses (Fig. 1). In addition, given that uncontrolled pulmonary inflammation is likely a leading cause of fatality in SARS-CoV-2 infection, we also attempt to speculate possible therapeutic interventions that may be applied to attenuate inflammatory responses in order to reduce mortality (Fig. 2).

Possible mechanisms of SARS-CoV-2-mediated inflammatory responses. Based on previous studies of SARS-CoV, we separate the inflammatory responses in SARS-CoV-2 infection into primary and secondary responses. Primary inflammatory responses occur early after viral infection, prior to the appearance of neutralizing antibodies (NAb). These responses are mainly driven by active viral replication, viral-mediated ACE2 downregulation and shedding, and host anti-viral responses. Secondary inflammatory responses begin with the generation of adaptive immunity and NAb. The virus-NAb complex can also trigger FcR-mediated inflammatory responses and acute lung injury.

Fc receptor-mediated antibody-dependent enhancement (ADE) of viral infection and inflammatory responses. A ADE occurs when antiviral neutralizing antibodies cannot completely neutralize the virus. Instead, the virus-NAb complex attaches to the Fc receptor (FcR), leading to viral endocytosis and infection of the target cells. The outcome is an increase in the overall replication of the virus and greater disease severity. B Virus-NAb complex binding to FcR can also activate proinflammatory signaling, skewing macrophage responses to the accumulation of proinflammatory (M1 or classically activated) macrophages in lungs. The M1 macrophages secrete inflammatory cytokines such as MCP-1 and IL-8, leading to lung injury. C Potential therapeutics based on targeting the Fc receptors to block SARS-CoV-2-induced inflammatory responses. From left to right, FcR can be blocked using anti-Fc specific antibodies, small molecules, or intravenous immunoglobulin (IVIG). The inhibitory FcR, FcγRIIB, may also be targeted to inhibit FcR activation. The FcRn can also be blocked by specific antibodies or inhibited competitively through IVIG binding.
Inflammation Caused by Rapid Viral Replication and Cellular Damage
Previous studies have shown that SARS-CoV predominantly infects airway and alveolar epithelial cells, vascular endothelial cells, and macrophages. In addition, SARS-CoV viral particles and viral genome have been detected in monocytes and lymphocytes (Gu et al.2005). SARS-CoV-2 uses the same entry receptor, angiotensin-converting enzyme 2 (ACE2), as SARS-CoV for infection, suggesting the likelihood of the same set of cells being targeted and infected (Zhao et al.2020; Zhou et al.2020). The early onset of rapid viral replication may cause massive epithelial and endothelial cell apoptosis and vascular leakage, triggering the release of exuberant pro-inflammatory cytokines and chemokines (Yang 2020). In addition, SARS-CoV-2 infection may also cause pyroptosis in macrophages and lymphocytes (Yang 2020). A vast majority of patients (82.1%) have been found to experience SARS-CoV-2-induced peripheral blood lymphopenia (Guan et al.2020), suggesting possible pulmonary infiltration of lymphocytes and/or cell damage through apoptosis or pyroptosis (Huang et al.2020). In SARS-CoV infection, viroporin 3a has also been shown to trigger the activation of NLRP3 (NOD-like receptor protein 3) inflammasome and the secretion of IL-1β in bone marrow-derived macrophages, suggesting the induction of cell pyroptosis (Chen et al.2019), which can cause the release of large amounts of proinflammatory factors (Fink and Cookson 2005).
Inflammation Caused by Virus-Induced ACE2 Downregulation and Shedding
ACE2-associated lung injury has been suggested in SARS-CoV infection (Imai et al.2008; Kuba et al.2005); SARS-CoV S protein can downregulate ACE2 (Glowacka et al.2010; Wang et al.2008), and induce the shedding of catalytically active ACE2 ectodomain (Haga et al.2008; Jia et al.2009; Lambert et al.2005). Loss of pulmonary ACE2 function has been suggested to be associated with acute lung injury (Imai et al.2005, 2008; Kuba et al.2005, 2006); the reduction in ACE2 function can cause dysfunction of the renin-angiotensin system (RAS) and enhance inflammation and vascular permeability. In a murine ARD model, loss of ACE2 expression resulted in enhanced vascular permeability, increased lung edema, neutrophil accumulation, and diminished lung function (Imai et al.2005). In addition, in human airway epithelia, ACE2 is constitutively shed by the action of disintegrin and metalloprotease 17 (ADAM17, also known as TNF-α cleavage enzyme, TACE) to release enzymatically active soluble ACE2 (sACE2) (Lambert et al.2005). Both SARS-CoV infection and inflammatory cytokines such as IL-1β and TNF-α can enhance ACE2 shedding (Haga et al.2008; Jia et al.2009; Lambert et al.2005). The biological function of sACE2 remains largely unknown. However, SARS-CoV S protein-induced ACE2 shedding has been found to be tightly coupled with TNF-α production in cell culture conditions (Haga et al.2008). Intriguingly, the S protein from another coronavirus, HNL63-CoV, does not induce ACE2 shedding, although the virus also binds to ACE2 to mediate NHL63-CoV entry (Haga et al.2008). HNL63-CoV infection only causes the common cold, suggesting a potential pathogenic role of sACE2 in SARS-CoV infection. These previous studies suggest that sACE2 may be directly involved in the inflammatory responses of SARS-CoV, and possibly SARS-CoV-2 as well.
Inflammatory Responses Induced by Anti-Spike IgG (Anti-S-IgG)
Antiviral neutralizing antibodies play an important role in viral clearance. However, previous studies in animal models have shown that in SARS-CoV infection, such anti-S protein-neutralizing antibodies (anti-S-IgG) can also cause severe lung injury by altering inflammatory responses (Liu et al.2019). In SARS-CoV/macaque models, it has been found that S-IgG present in infected lungs can facilitate severe lung injury; in these SARS-CoV S protein-vaccinated Chinese macaques, acute lung injury was more pronounced than in unvaccinated control animals that showed only minor to moderate lung inflammation (Liu et al.2019). Consistent with this observation, adoptive transfer of purified anti-S-IgG-neutralizing antibody (i.v. injection) to macaques, despite the fact that it reduced viral loads following subsequent challenge with SARS-CoVPUMC, led to acute diffuse alveolar damage in all infected animals, whereas in the control group (injected with non-specific IgG), only minor to moderate inflammation in the lungs was observed (Liu et al.2019). This animal study suggests that despite viral suppression, the presence of anti-spike protein antibody at the acute stage of SARS-CoV infection can actually cause severe acute lung injury that persists until the late stages. Similar observations of SARS-CoV vaccine-induced pulmonary injury have also been reported in multiple animal models using mice and African green monkeys (Bolles et al.2011; Clay et al.2012; Tseng et al.2012).
Results from these animal studies also appear to mirror some of the clinical observations in SARS-CoV infected patients: the development of acute respiratory disease coincides with antiviral IgG seroconversion in 80% of patients (Peiris et al.2003). In addition, it was found that patients who developed the anti-S-neutralizing antibody faster had a higher chance of dying from the disease; it took an average of only 14.7 days for the deceased patients to reach their peak levels of neutralizing antibody activities, as opposed to 20 days for the recovered patients (Zhang et al.2006).
The mechanisms of the anti-S neutralizing antibody-induced inflammation and lung injury remain only partially understood. It has been proposed that the presence of S-IgG prior to viral clearance alters the functional polarization of alveolar macrophages in acutely infected macaques. Anti-S-IgG can promote proinflammatory monocyte/macrophage accumulation and the production of MCP-1 and IL-8 in the lungs. Such anti-S-IgG-initiated proinflammatory responses appear to be mediated through the binding of the virus-anti-S-IgG complex to the Fc receptors (FcR) present on monocytes/macrophages, as FcR blockade reduces the production of inflammatory cytokines (Liu et al.2019). It is also possible that such a virus-anti-S-IgG complex may additionally activate the classical pathway of the complement system, leading to cellular damages, although this has not been investigated. Alternatively, antibody-dependent cell-mediated cytotoxicity (ADCC) may also be involved.
A major question in SARS-CoV-induced pulmonary disease is why a small percentage of patients, particularly those who produce neutralizing antibody early, experience persistent inflammation, ARD, and eventually succumb, while other patients survive the inflammatory responses and clear the virus. We speculate that a possible underlying mechanism may be related to antibody-dependent enhancement (ADE) of viral infection that occurs in some patients with early, sub-optimal antibody activity that cannot completely clear the virus, but instead leads to persistent viral replication and inflammation (Fig. 2). ADE is a well-known virology phenomenon that has been demonstrated in the infections of multiple viruses such as dengue, flavivirus, and influenza virus (Halstead and O’Rourke 1977; Haslwanter et al.2017; Ochiai et al.1992; Takada et al.2003; Takada and Kawaoka 2003). ADE promotes viral cellular uptake of infectious virus-antibody complexes following their interaction with FcR or other receptors, leading to enhanced infection of target cells (Halstead and O’Rourke 1977; Haslwanter et al.2017; Ochiai et al.1992; Takada et al.2003; Takada and Kawaoka 2003). Thus, interaction of FcR with the virus-anti-S-IgG complex may facilitate both inflammatory responses and persistent viral replication in the lungs of some patients (Fig. 2).
Given that there are very few mechanistic studies on inflammatory responses in SARS-CoV infection, we focus only on discussing limited mechanisms that might be involved. For the convenience of further discussion of potential therapeutics, here we separate SARS-CoV-mediated inflammatory responses into two different stages (Fig. 1): the primary response and the secondary response. Primary inflammatory responses occur early after viral infection, prior to the appearance of neutralizing antibodies (NAb). These responses are mainly driven by active viral replication, viral-mediated ACE2 downregulation and shedding, and host antiviral responses, which can lead to increased cytokine/chemokine production and cellular damage through apoptosis and/or pyroptosis. Most patients can tolerate primary inflammatory responses with a positive outcome of viral load reduction or even viral clearance, followed by receding of inflammation. Secondary inflammatory responses begin with the generation of adaptive immunity and the appearance of NAb that further diminish viral replication. However, as described above, the appearance of NAb can also trigger FcR-mediated inflammatory responses and cause severe lung injury. In SARS-CoV infected patients, the appearance of antiviral IgG coincides with the onset of acute respiratory disease in 80% of patients (Peiris et al.2003). A possible underlying mechanism is likely antibody-dependent enhancement (ADE) of viral infection that leads to persistent viral replication and inflammatory responses from macrophages.
Given that most patients can survive primary inflammatory responses, we mainly focus on discussing secondary inflammatory responses that frequently lead to fatality. There are several potential therapeutic approaches that may be applied or developed (Fig. 2). These approaches focus primarily on blocking FcR receptor to prevent virus-NAb complex binding to FcR to trigger inflammatory responses (Nimmerjahn and Ravetch 2008a, c). First, FcR can be blocked by specific antibodies to inhibit its activation. Small-molecule inhibitors can also be developed to interact with the Ig-binding domains of FcR to block FcR activation. Second, the inhibitory FcR, FcγRIIB, may also be targeted to inhibit FcR activation. Several FcγRIIB specific antibodies are now being developed for potential use as novel immune suppressors (van Mirre et al.2004; Veri et al.2007). Third, FcR activation can also be inhibited by targeting the neonatal Fc receptor (FcRn), which is essential for extending the half-life of IgG. Antibody or small molecule-mediated blockage of FcRn can prevent IgG interaction with FcRn, which can decrease circulating IgG levels (Nimmerjahn and Ravetch 2008b). In addition, intravenous immunoglobulin (IVIG) can be used to saturate the IgG recycling capacity of FcRn and proportionately reduce the levels of antiviral NAb. IVIG can also competitively block the binding of antiviral NAb to other FcRs (Kurlander and Hall 1986).
Although we mainly focus on strategies targeting virus-NAb complex binding to FcR, it is possible that the virus-NAb complex may also initiate inflammatory responses through the classical pathway of the complement system, which may be blocked through C5- and C5a-targeted inhibition (Horiuchi and Tsukamoto 2016).
In sum, at present, there is no effective antiviral therapy for SARS-CoV-2 infection, which frequently leads to fatal inflammatory responses and acute lung injury. Here, we discuss the various mechanisms of SARS-CoV-mediated inflammation. We also assume that SARS-CoV-2 likely shares similar inflammatory responses. Potential therapeutic tools to reduce SARS-CoV-2-induced inflammatory responses include various methods to block FcR activation. In the absence of a proven clinical FcR blocker, the use of intravenous immunoglobulin to block FcR activation may be a viable option for the urgent treatment of pulmonary inflammation to prevent severe lung injury. Such treatment may also be combined with systemic anti-inflammatory drugs or corticosteroids. However, these strategies, as proposed here, remain to be clinically tested for effectiveness.
References
Bolles M, Deming D, Long K, Agnihothram S, Whitmore A, Ferris M, Funkhouser W, Gralinski L, Totura A, Heise M, Baric RS (2011) A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J Virol 85:12201–12215
Chen IY, Moriyama M, Chang MF, Ichinohe T (2019) Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front Microbiol 10:50
Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, Qiu Y, Wang J, Liu Y, Wei Y, Xia J, Yu T, Zhang X, Zhang L (2020) Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet. https://doi.org/10.1016/S0140-6736(20)30211-7
Clay C, Donart N, Fomukong N, Knight JB, Lei W, Price L, Hahn F, Van Westrienen J, Harrod KS (2012) Primary severe acute respiratory syndrome coronavirus infection limits replication but not lung inflammation upon homologous rechallenge. J Virol 86:4234–4244
Fink SL, Cookson BT (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916
Glowacka I, Bertram S, Herzog P, Pfefferle S, Steffen I, Muench MO, Simmons G, Hofmann H, Kuri T, Weber F, Eichler J, Drosten C, Pöhlmann S (2010) Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J Virol 84:1198–1205
Gu J, Gong E, Zhang B, Zheng J, Gao Z, Zhong Y, Zou W, Zhan J, Wang S, Xie Z, Zhuang H, Wu B, Zhong H, Shao H, Fang W, Gao D, Pei F, Li X, He Z, Xu D, Shi X, Anderson VM, Leong AS (2005) Multiple organ infection and the pathogenesis of SARS. J Exp Med 202:415–424
Guan WJ, Ni ZY, Hu Y, Liang WH, Ou C, He J, Liu L, Shan H, Lei C, Hui DSC et al (2020) Clinical characteristics of 2019 novel coronavirus infection in China. MedRxiv. https://doi.org/10.1101/2020.02.06.20020974
Haga S, Yamamoto N, Nakai-Murakami C, Osawa Y, Tokunaga K, Sata T, Yamamoto N, Sasazuki T, Ishizaka Y (2008) Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc Natl Acad Sci U S A 105:7809–7814
Halstead SB, O’Rourke EJ (1977) Antibody-enhanced dengue virus infection in primate leukocytes. Nature 265:739–741
Haslwanter D, Blaas D, Heinz FX, Stiasny K (2017) A novel mechanism of antibody-mediated enhancement of flavivirus infection. PLoS Pathog 13:e1006643
Horiuchi T, Tsukamoto H (2016) Complement-targeted therapy: development of C5- and C5a-targeted inhibition. Inflamm Regen 36:11
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, Cheng Z, Yu T, Xia J, Wei Y, Wu W, Xie X, Yin W, Li H, Liu M, Xiao Y, Gao H, Guo L, Xie J, Wang G, Jiang R, Gao Z, Jin Q, Wang J, Cao B (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan. Lancet China. https://doi.org/10.1016/S0140-6736(20)30183-5
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui CC, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116
Imai Y, Kuba K, Penninger JM (2008) The discovery of angiotensin-converting enzyme 2 and its role in acute lung injury in mice. Exp Physiol 93:543–548
Jia HP, Look DC, Tan P, Shi L, Hickey M, Gakhar L, Chappell MC, Wohlford-Lenane C, McCray PB Jr (2009) Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol 297:L84–L96
Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11:875–879
Kuba K, Imai Y, Penninger JM (2006) Angiotensin-converting enzyme 2 in lung diseases. Curr Opin Pharmacol 6:271–276
Kurlander RJ, Hall J (1986) Comparison of intravenous gamma globulin and a monoclonal anti-Fc receptor antibody as inhibitors of immune clearance in vivo in mice. J Clin Invest 77:2010–2018
Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM, Turner AJ (2005) Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem 280:30113–30119
Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, Tang H, Nishiura K, Peng J, Tan Z, Wu T, Cheung KW, Chan KH, Alvarez X, Qin C, Lackner A, Perlman S, Yuen KY, Chen Z (2019) Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. https://doi.org/10.1172/jci.insight.123158
Nimmerjahn F, Ravetch JV (2008a) Analyzing antibody-Fc-receptor interactions. Methods Mol Biol 415:151–162
Nimmerjahn F, Ravetch JV (2008b) Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol 26:513–533
Nimmerjahn F, Ravetch JV (2008c) Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 8:34–47
Ochiai H, Kurokawa M, Matsui S, Yamamoto T, Kuroki Y, Kishimoto C, Shiraki K (1992) Infection enhancement of influenza A NWS virus in primary murine macrophages by anti-hemagglutinin monoclonal antibody. J Med Virol 36:217–221
Peiris JS, Chu CM, Cheng VC, Chan KS, Hung IF, Poon LL, Law KI, Tang BS, Hon TY, Chan CS, Chan KH, Ng JS, Zheng BJ, Ng WL, Lai RW, Guan Y, Yuen KY, HKU/UCH SARS Study Group (2003) Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361:1767–1772
Takada A, Kawaoka Y (2003) Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol 13:387–398
Takada A, Feldmann H, Ksiazek TG, Kawaoka Y (2003) Antibody-dependent enhancement of Ebola virus infection. J Virol 77:7539–7544
Tseng CT, Sbrana E, Iwata-Yoshikawa N, Newman PC, Garron T, Atmar RL, Peters CJ, Couch RB (2012) Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 7:e35421
van Mirre E, van Royen A, Hack CE (2004) IVIg-mediated amelioration of murine ITP via FcgammaRIIb is not necessarily independent of SHIP-1 and SHP-1 activity. Blood 103:1973; author reply 1974
Veri MC, Gorlatov S, Li H, Burke S, Johnson S, Stavenhagen J, Stein KE, Bonvini E, Koenig S (2007) Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology 121:392–404
Wang S, Guo F, Liu K, Wang H, Rao S, Yang P, Jiang C (2008) Endocytosis of the receptor-binding domain of SARS-CoV spike protein together with virus receptor ACE2. Virus Res 136:8–15
Wong CK, Lam CW, Wu AK, Ip WK, Lee NL, Chan IH, Lit LC, Hui DS, Chan MH, Chung SS, Sung JJ (2004) Plasma inflammatory cytokines and chemokines in severe acute respiratory syndrome. Clin Exp Immunol 136:95–103
Yang M (2020) Cell pyroptosis, a potential pathogenic mechanism of 2019-nCoV infection. SSRN. https://doi.org/10.2139/ssrn.3527420
Zhang L, Zhang F, Yu W, He T, Yu J, Yi CE, Ba L, Li W, Farzan M, Chen Z, Yuen KY, Ho D (2006) Antibody responses against SARS coronavirus are correlated with disease outcome of infected individuals. J Med Virol 78:1–8
Zhao Y, Zhao Z, Wang Y, Zhou Y, Ma Y, Zou W (2020) Single-Cell RNA expression profiling of ACE2, the putative receptor of Wuhan 2019-nCoV. BioRxiv. https://doi.org/10.1101/2020.01.26.919985
Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, Guo H, Jiang RD, Liu MQ, Chen Y, Shen XR, Wang X, Zheng XS, Zhao K, Chen QJ, Deng F, Liu LL, Yan B, Zhan FX, Wang YY, Xiao GF, Shi ZL (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. https://doi.org/10.1038/s41586-020-2012-7
Acknowledgments
The authors wish to thank Feng Li for helpful discussions and Jennifer Guernsey for editorial assistance.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Animal and Human Rights Statement
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
About this article

Cite this article
Fu, Y., Cheng, Y. & Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 35, 266–271 (2020). https://doi.org/10.1007/s12250-020-00207-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-020-00207-4