
Abstract
Thirty-one proteins are known to form extracellular fibrillar amyloid in humans. Molecular information about many of these proteins in their monomeric, intermediate or fibrillar form and how they aggregate and interact to form the insoluble fibrils is sparse. This is because amyloid proteins are notoriously difficult to study in their soluble forms, due to their inherent propensity to aggregate. Using recent developments in fast NMR techniques, band-selective excitation short transient and band-selective optimized flip-angle short-transient heteronuclear multiple quantum coherence we have been able to assign a 5 kDa full-length amyloidogenic protein called medin. Medin is the key protein component of the most common form of localised amyloid with a proposed role in aortic aneurysm and dissection. This assignment will now enable the study of the early interactions that could influence initiation and progression of medin aggregation. The chemical shifts have been deposited in the BioMagRes-Bank accession Nos. 25399 and 26576.
Similar content being viewed by others


Biological context
Medin is the principal protein component of the most common form of localised amyloid and is thought to have a role in aortic aneurysm and dissection. Despite the prevalence of these amyloid plaques, medin is a relatively poorly understood protein. Medin is proposed to derive from the cleavage of the milk-fat globule protein MFG-E8. The medin sequence is located within the C2 domain of MFG-E8 but the mechanism for excision is not understood (Häggqvist et al. 1999). Within the PDB there are three deposited structures of the C2 domain of MFG-E8; two bovine structures determined by X-ray crystallography (2PQS and 3BN6) and a murine structure determined by solution state NMR (2LDL). There is only 66 % sequence identity between murine and human and 69 % between bovine and human C2 domains. To our knowledge there are no assignments or structures of the human form of MFG-E8 or any isolated domain structures. Previous biophysical measurements of medin at 20 μM concentration indicated that fibrillisation occurred after approximately 30 h (Davies et al. 2014, 2015). Traditional NMR assignment methods typically require high protein concentrations. Due to the nucleation dependent growth of amyloid proteins, increased protein concentration can reduce the lag time for fibrillisation (Harper and Lansbury 1997), making time-consuming assignment experiments of these proteins in their soluble form prohibitive. As an alternative we sought to assign medin under denaturing conditions (8 M Urea) and subsequently transfer the assignment to spectra collected on the initial pre-fibrillar native form. This enabled an increase in protein concentration and reduced peak-width, essential for collecting the less sensitive triple resonance NMR experiments. Additionally the use of fast pulsing (BEST) multidimensional NMR experiments enabled triple resonance experiments to be collected in a fraction of the time.
Methods and experiments
13C, 15N isotope labelled medin were expressed as previously described (Davies et al. 2014) with the substitution of Lemo 21 (DE3) cells. Cell pellets were resuspended in 6 M guanidine hydrochloride (GdmCl), 20 mM sodium phosphate, 0.5 M NaCl, pH 8.0 and frozen at −20 °C. Cells were thawed, homogenised, and cell debris removed by centrifugation (19,000g, 15 min, 4 °C). The supernatant was loaded onto a 5 ml Ni2+–NTA column and washed with 4 column volumes (CV) of 6 M GdmCl, pH 8, followed by 4 CV of 6 M GdmCl, pH 6, and eluted with 3 CV of 6 M GdmCl, pH 2, and stored at −20 °C. Fusion protein was buffer exchanged into 20 mM Tris–Cl, 0.15 mM NaCl, pH 7.4, and the His6-SUMO tag was removed by incubation with SUMO protease I at 4 °C for 3 h. The cleavage mixture was then passed through a 5 ml Ni2+–NTA column and the flow through containing medin collected. Medin was buffer exchanged into d dH2O and lyophilised. The lyophilised protein was then resuspended in 20 mM sodium phosphate, 20 mM NaCl, pH 6.5 with or without the addition of 8 M urea for NMR assignment. For triple resonance assignment medin was reconstituted to a concentration of 200 μM in 8 M urea with 10 % (v/v) 2H2O. Spectra were acquired at 25 °C on Bruker AVANCE III 600 and 800 MHz spectrometers equipped with 5 mm triple resonance (TCI) cryoprobes. A schematic of the experimental workflow is detailed in Fig. 1a. In brief, 2D SOFAST-HMQC and 3D BEST-experiments (HNCO, HNCACO, HNCOCACB, HNCACB) were recorded for 1HN, 13Cα, 13Cβ 13C′ and 15NH assignments. Spectra were assigned in 8 M urea and the NH and HN backbone assignment then transferred using a series of 2D HSQC experiments collected at 6, 4, 2, 1 and 0 M urea. Spectra were processed using Topspin 3.1 (Bruker) and assignment carried out using CCPN Analysis (Vranken et al. 2005). SOFAST-HMQC experiments were interleaved throughout to assess sample stability.

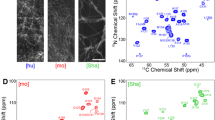
NMR assignment of medin. An iterative process was undertaken to achieve assignment of medin in a physiological buffer, utilising denaturing conditions and fast-pulsing NMR techniques (a). Assigned 1H–15N HSQC for medin in a physiological buffer (20 mM sodium phosphate, 20 mM NaCl, pH 6.5) (b). Spectrum collected with 40 μM 15N uniformly labelled medin sample
Assignment and data deposition
1H–15N HSQC spectrum exhibited peaks consistent with the protein being predominantly unfolded and gave discrete peaks of limited dispersion. In 8 M urea, medin remained in solution for at least 60 h at 200 μM. 3D sequential assignment experiments were performed using BEST experiments to increase the number of scans attainable during the stable phase of the protein (Schanda et al. 2006; Lescop et al. 2007). As a result, a complete 15N, 1HN, assignment of the stable denatured state was achieved (excluding N and C-terminal residues), along with over 89 % for backbone atoms 13CO, 13Cα, 13Cβ. The corresponding chemical shifts have been deposited in the BioMagRes-Bank; Accession No. 25399. A urea titration series (8–0 M) was used to transfer assignments to a 1H–15N HSQC spectrum with 0 M urea, the corresponding chemical shifts have been deposited in the BioMagRes-Bank; Accession No. 26576. To remove ambiguity in some peak assignments, BEST experiments were also carried out in 4 M urea. The interleaved SOFAST-HMQC data were acquired in 8 min (Schanda and Brutscher 2005; Schanda et al. 2005). Triple resonance spectra were acquired for soluble medin in 8 M urea in 26 h with resolution in the indirect dimension high enough to resolve all unfolded signals. Urea titration enabled transfer of NH assignment to the native form resulting in 94 % of backbone NH shifts assigned in 0 M urea (Fig. 1b). Chemical shift changes from 8 M to 0 M urea were systematic and rarely deviated in the 15N dimension indicative of minimal conformational changes and also suggesting that medin has a predominantly random coil structure in 0 M urea.
This method of assignment transfer utilizing a combination of fast pulsing techniques and denaturants sufficiently extended the duration of protein in the soluble phase (although it should be noted the presence of denaturant did not halt the process of aggregation completely). We envisage that this method of assignment can be used for other rapidly aggregating proteins that would be otherwise intractable to NMR assignment in the soluble phase.
References
Davies HA, Wilkinson MC, Gibson RP, Middleton DA (2014) Expression and purification of the aortic amyloid polypeptide medin. Protein Expr Purif 98:32–37
Davies HA, Madine J, Middleton DA (2015) Comparisons with amyloid-β reveal an aspartate residue that stabilizes fibrils of the aortic amyloid peptide medin. J Biol Chem 290:7791–7803
Häggqvist B, Näslund J, Sletten K, Westermark GT, Mucchiano G, Tjernberg LO, Nordstedt C, Engström U, Westermark P (1999) Medin: an integral fragment of aortic smooth muscle cell-produced lactadherin forms the most common human amyloid. Proc Natl Acad Sci 96(15):8669–8674
Harper JD, Lansbury PT (1997) Models of amyloid seeding in Alzheimier’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 66:385–407
Lescop E, Schanda P, Brutscher B (2007) A set of BEST triple-resonance experiments for time-optimized protein resonance assignment. J Magn Reson 187(1):163–169
Schanda P, Brutscher B (2005) Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc 127(22):8014–8015
Schanda P, Kupče Ē, Brutscher B (2005) SOFAST-HMQC experiments for recording two-dimensional deteronuclear correlation spectra of proteins within a few seconds. J Biomol NMR 33(4):199–211
Schanda P, Van Melckebeke H, Brutscher B (2006) Speeding up three-dimensional protein NMR experiments to a few minutes. J Am Chem Soc 128(28):9042–9043
Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas M, Ulrich EL, Markley JL, Ionides J, Laue ED (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins: Struct, Funct, Bioinf 59(4):687–696
Acknowledgments
Financial support for this work was provided by the British Heart Foundation (JM/HAD, FS/12/61/29877). This work was supported by an award from the University of Liverpool Technology Directorate Voucher Scheme and the NMR Centre for Structural Biology. We thank Professor Lu-Yun Lian for discussion and advice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Davies, H.A., Phelan, M.M. & Madine, J. 1H, 15N and 13C assignment of the amyloidogenic protein medin using fast-pulsing NMR techniques. Biomol NMR Assign 10, 75–77 (2016). https://doi.org/10.1007/s12104-015-9641-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12104-015-9641-z