
Abstract
Balanced signal transduction is crucial in tissue patterning, particularly in the vasculature. Heterotopic ossification (HO) is tightly linked to vascularization with increased vessel number in hereditary forms of HO, such as Fibrodysplasia ossificans progressiva (FOP). FOP is caused by mutations in the BMP type I receptor ACVR1 leading to aberrant SMAD1/5 signaling in response to ActivinA. Whether observed vascular phenotype in human FOP lesions is connected to aberrant ActivinA signaling is unknown. Blocking of ActivinA prevents HO in FOP mice indicating a central role of the ligand in FOP. Here, we established a new FOP endothelial cell model generated from induced pluripotent stem cells (iECs) to study ActivinA signaling. FOP iECs recapitulate pathogenic ActivinA/SMAD1/5 signaling. Whole transcriptome analysis identified ActivinA mediated activation of the BMP/NOTCH pathway exclusively in FOP iECs, which was rescued to WT transcriptional levels by the drug candidate Saracatinib. We propose that ActivinA causes transcriptional pre-patterning of the FOP endothelium, which might contribute to differential vascularity in FOP lesions compared to non-hereditary HO.
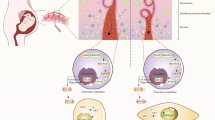
Graphical abstract

Similar content being viewed by others


Introduction
The vasculature is a complex, dynamic network of branching vessels lined by endothelial cells (ECs). Controlled blood vessel formation is crucial in embryogenesis and adult tissue homeostasis, requiring proper vascular patterning coordinated by multiple signaling cascades, including vascular endothelial growth factor (VEGF), NOTCH and Bone morphogenetic protein (BMP) pathways [1]. Aberrant activation of signaling pathways causes vascular malformations as observed in pathological conditions such as cancer and chronic inflammation [2]. The pathology of heterotopic ossification (HO) is defined by ectopic bone formation within soft tissues and is the main clinical symptom in rare hereditary forms of HO, but also a common issue of trauma and surgery [3]. Pre-osseous lesions are highly angiogenic and fibroproliferative, followed by an avascular chondrogenic stage and subsequent formation of mature, vascularized heterotopic bone through endochondral ossification [4,5,6]. Thus, blood vessels in HO undergo rapid and dynamic changes, but it remains elusive how signaling molecules orchestrate the vasculature in the aberrant tissue repair processes. It was recently shown that blockage of pro-angiogenic VEGFA reduced trauma-induced HO, highlighting vascularization as a therapeutic target [7].
Interestingly, human HO biopsies uncovered a differential vascular phenotype with increased vessel number, area and size in genetic versus non-hereditary forms distinguishing both pathologies [8]. Fibrodysplasia ossificans progressiva (FOP), a hereditary form of HO, is caused by gain of function mutations in the BMP type 1 receptor ACVR1 (ALK2) with R206H being the most common point mutation located in the intracellular glycine-serine (GS) rich domain [9]. Mutant receptors lead to hyperactivated SMAD1/5 signaling in response to BMPs [10] and aberrantly transduce SMAD1/5 signaling in response to ActivinA [11, 12]. Activins are TGF-β family members and normally signal via the type I receptors ACVR1B/C, intracellularly activating SMAD2/3 [13]. In FOP mice, blocking of ActivinA prevents HO indicating a central role of this ligand in the disease [11]. Whether ActivinA causes the vascular phenotype observed in human FOP biopsies is unknown.
ECs derived from induced pluripotent stem cells (iPSC), here called iECs, are an attractive in vitro model of the human endothelium. To date it is unclear whether iECs recapitulate primary ECs from FOP patients and are responsive to ActivinA. Current literature suggests that FOP iECs have reduced viability [14] and do not show aberrant ActivinA/SMAD1/5 signaling [15].
Here, we optimized iPSC differentiation conditions and generated a new iEC FOP model to investigate the effect of ActivinA on ACVR1 signaling in FOP iECs to better understand the underlying molecular mechanisms of the vascular phenotype. We demonstrate aberrant ActivinA/SMAD1/5 signaling and a unique transcriptome in FOP iECs interlinking ActivinA with BMP/NOTCH pathway activation. Moreover, we show that the drug candidate Saracatinib rescued the ActivinA-induced transcriptome in FOP iECs to WT levels suggesting a preventive effect on aberrant vascularization in early HO lesions in FOP.
Materials and Methods
iPSC Cell Culture
Cell lines iPSC-WT-1 (BCRTi005-A), iPSC-WT-2 (BCRTi004-A), iPSC-FOP-1 (BCRTi001-A), iPSC FOP-2 (BCRTi002-A) were generated from urinary cells as described previously [16,17,18]. All lines are registered, including ethical statements at the global hiPSC registry https://hpscreg.eu. iPSCs were cultured in defined conditions in E8 medium [19] in Geltrex-coated 6-well plates and routinely passaged at a ratio of ~1:20 every 4–5 days using 0.5 mM EDTA.
Differentiation of iECs from iPSCs
Cryopreserved iPSCs (according to the CryoPause method in a controlled-rate freezer [20]), were thawed and immediately differentiated into iECs using a modified version of [21]. In brief, iPSCs were thawed in E8 medium supplemented with 10 μM ROCK inhibitor (Y-27632; Stem Cell Technologies, 72,305) and seeded at a density of 2–3 × 105 cells/well on Geltrex coated 6-well plates. About 24 h after seeding the medium was changed to mesoderm induction medium containing N2B27 medium (1:1 mix of Neurobasal and DMEM/F12 with GlutaMAX and supplemented with N2 and B27 (−) Vitamin A (all Thermo Fisher), with 25 ng/mL BMP4 (PeproTech) and 6-7 μM CHIR99021 (BioVision). On day 4 the medium was changed to iEC induction medium consisting of StemPro-34 (Thermo Fisher) supplemented with 200 ng/mL VEGFA165 (PeproTech) and 2 μM Forskolin (Abcam). The iEC induction medium was renewed on day 5. On day 6 the EC fraction was measured by FACS CD144-FITC and sorted by MACS using CD144 MicroBeads (Miltenyi Biotec). Upon collection of the negative fraction, the positive fraction (CD144+) was eluted. Cells were resuspended in Expansion Medium [22] (EGM-2 containing single quots, w/o Hydrocortisone (LONZA) supplemented with 20% FCS, 100 units/ml penicillin, 10 μg/ml streptomycin (PAA-laboratories), 10 μM SB431542 (Selleck Chemicals)) and 4 × 104 cells per cm2 were seeded on 0.1% gelatine (EmbryoMax, Sigma-Aldrich) coated culture flasks. Medium change was performed the next day and subsequently every 2–3 days. Cells were split (max. 1:3) or frozen (90% FCS, 10% DMSO) upon confluency. Upon thawing iECs were cultured in Growth Medium (EGM2 containing single quots, w/o Hydrocortisone (PromoCell) supplemented with 20% FCS, 100 units/ml penicillin, 10 μg/ml streptomycin) on 0.1% gelatin coated culture flasks and used for all experiments in passage 2.
Cell Stimulation with Growth Factors and SMKI Treatment
Cells were starved prior growth factor stimulation for 4 h. iPSC were starved in Essential 6 medium (Thermo Fisher) and iECs in Endothelial Basal Medium 2 (EBM2) (PromoCell) supplemented with 0.5% FCS and 100 units/ml penicillin, 10 μg/ml streptomycin (PAA Laboratories). Small-molecule kinase inhibitors (SMKI) were added to cells 1 h prior ligand stimulation with indicated concentrations unless stated otherwise. Growth factors and SMKIs were reconstituted and stored according to manufacturer instructions. hBMP6 (S. Vukicevic, University of Zagreb, Croatia), rhActivin-A (R&D Systems), hTNF-α (ImmunoTools), Saracatinib, K02288, SB431542 (Selleck Chemicals).
Adhesion Molecule Expression Assay
iECs were exposed to tumor necrosis factor-alpha (TNF-α) (0.6 nM) for 2 h. RNA was isolated and expression of ICAM-1 was analyzed by qPCR.
Barrier Function
Endothelial barrier function was assessed by Electric Cell-substrate Impedance Sensing (ECIS) using the ECIS Zθ instrument (Applied BioPhysics, ibidi). iECs were grown to confluence on 0.1% gelatine (EmbryoMax, Sigma Aldrich) coated 8W10E arrays (ibidi) (3 × 10 4 cells/ well) for 72 h. Cells were starved for 5 h before stimulation with VEGFA (5 nM) and PBS as control. Barrier function was assessed by ECIS measurement of resistance at 4000 Hz.
Tube Formation Assay
iECs were seeded in growth medium on growth factor reduced Matrigel (Corning) coated 96 well culture plates (3 × 104/well). After an incubation for 24 h at 37 °C in 5% CO2 images were taken using phase-contrast microscopy.
Western Blot Analysis
Protein lysates were separated on 10% SDS-PAGE gels. Gels were transferred to PVDF membranes by Western blotting. Membranes were blocked in 0.1% TBS-T containing 3% w/v BSA for 1 h at RT, washed three times in 0.1% TBS- T and incubated with indicated primary antibodies (GAPDH #2118; phospho-SMAD1/5 (Ser463/465) #9516; SMAD1 #6944; phospho-SMAD2 (Ser465/467) #3108, SMAD2 #3122, Cell Signaling Technology) overnight at 4 °C following manufacturer’s instructions. Secondary HRP conjugated antibodies goat-anti-mouse or goat-anti-rabbit IgG (Dianova) were used. Chemiluminescent reactions were processed using WesternBright Quantum HRP substrate (advansta) and documented by using a ChemiSmart5000 digital imaging system (Vilber-Lourmat).
qRT-PCR
Cells were washed once with DPBS and RNA was isolated using NucleoSpin RNA II (Macherey-Nagel) according to manufacturer instructions. The amount of 0.5–1 μg RNA was reverse transcribed into cDNA using M-MLV reverse transcriptase and random primers (NEB). qRT-PCR was performed using PCR Luna Universal qPCR Master Mix (NEB) and specific primers listed in Table 1. Expression levels were assessed by StepOne Plus, and StepOne Software 2.3 (Applied Biosystems) and measured in technical replicates. Target gene expression was quantified relative to the housekeeping gene RSP9 using the ΔΔCT method including primer efficiency [23].
Immunofluorescence Staining
3 × 104 cells were seeded on glass coverslips placed in 24-well plates until they formed a confluent monolayer. Cells were fixed with 4% paraformaldehyde (PFA), quenched in 50 mM ammonium chloride and permeabilized in 0.5% Triton-X-100 for 15 min. After blocking for 1 h in 3% w/v BSA and 5% v/v normal goat serum in PBS, cells were stained with primary antibodies (VE-Cadherin, #2500, 1:400; PECAM-1 #3528, 1:200; Cell Signaling Technology) in blocking solution overnight. Fluorophore conjugated secondary antibodies, (1:300) (Alexa Fluor™ 488 F(ab’)2-goat-α-mouse IgG (A-10684, Invitrogen) and Alexa Fluor™ 594 goat-α-rabbit IgG (A-11012, Invitrogen)), were diluted in blocking solution and incubated for 1 h at RT. Nuclei were stained with DAPI (Sigma-Aldrich) using a 1:1000 dilution in DPBS for 30 min at RT.
FACS Analysis
Antibodies for surface marker staining (CD144-FITC-human clone REA199, CD31-APC-human clone AC128 (Miltenyi Biotec)) were used according manufactures instructions. Measurement was performed by MACSQuant VYB (Miltenyi Biotec) and analyzed by FCS Express V6 software.
RNA-Seq Library Preparation and Sequencing
8 × 104 iECs per well were seeded in 12 well plate and grown to confluence generated from 4 biological independent iPSC lines. Two independent experiments of ligand and SMKI treatment were performed for each line. Upon starvation, ligand stimulation and SMKI treatment cells were lysed and RNA was isolated according to manufacturer instructions (Macherey-Nagel). RNA samples were sent for Sequencing to Genewiz, Leipzig, Germany.
RNA-Seq Data Analysis
The sequencing data was uploaded to the Galaxy web platform, and the public server at usegalaxy.eu was used to analyze the data [24]. Quality of raw reads was performed with FASTQC before data was mapped to the reference genome (hg38) using STAR mapper [25]. Alignment quality was assessed with MultiQC [26] and RNA-Seq alignments were assembled into potential transcripts by StringTie [27]. This output was used to analyze differential gene expression with DESeq2 [28]. Cutoff for differentially expressed genes was a logarithmic fold change of ≥0.58 and with an adjusted p value of 0.05. Shared differentially expressed genes in both FOP donors was assessed with BioVenn [29]. Z-score calculation and generation of heatmaps was performed with the „pheatmap “package in RStudio. Gene ontology was performed with DAVID Bioinformatic Resources 6.8 [30, 31] and applying Benjamini correction.
Statistical Analysis
All experiments of each of the four biological replicates (iPSC donors) was independently repeated at least three times. Statistical analysis and data illustration was performed with GraphPad Prism 8 (GraphPad Software Inc.). Normal distribution of data sets n < 5 were tested with the Shapiro-Wilk normality test. Data sets n ≥ 5 were tested additionally with the Kolmogorov Smirnov test for normality. In cases of failure to reject the null hypothesis, the ANOVA and Bonferroni post hoc test were used to check for statistical significance under the normality assumption. P-values lower than 0.05 were considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001).
Results
Optimized Differentiation Conditions to Generate Functional ECs from FOP iPSCs
In this study we aimed to investigate ActivinA signaling in FOP ECs. Due to a high risk of HO-induction in FOP patients by biopsy retrieval, the establishment of cell models has been challenging. iPSCs from two FOP patients (ACVR1 R206H) and two controls, which we characterized previously [16,17,18], were used to generate iECs (FOP-1, FOP-2, WT-1, WT-2) (Fig. S1a). All four iPSC lines responded to ActivinA with a dose-dependent increase in SMAD2 phosphorylation (pSMAD2) (Fig. 1a and S1b), while ActivinA-induced SMAD1/5 phosphorylation (pSMAD1/5) was only seen in FOP iPSCs (Fig. 1a and S1b). As a control, treatment with BMP6, an ACVR1 ligand, showed dose-dependent phosphorylation of SMAD1/5 in both, WT and FOP iPSCs (Fig. 1a and S1b). Based on aberrant ActivinA/SMAD1/5 signaling in FOP iPSCs, we optimized an EC differentiation protocol devoid of exogenous ActivinA [21] in combination with CryoPause [20] (Fig. 1b) by directly seeding iPSCs for differentiation after thawing. On day 1, mesoderm was induced by BMP4 and the GSK3-β inhibitor CHIR. On day 4, endothelial expansion of mesodermal cells was achieved by VEGFA and Forskolin. The EC fraction was measured by FACS and subsequently purified by MACS using vascular endothelial cadherin (VE-Cadherin/CD144) as a marker. WT and FOP iPSC differentiated to iECs with efficiencies up to ~80% (Fig. 1c). Both iECs form dense monolayers, express the junctional markers VE-Cadherin and PECAM-1 as assessed by immunofluorescence staining and FACS analysis (Fig.1d, e) and quantitative RT-PCR (Fig. 1g). iECs expressed additional EC markers, such as Vascular endothelial growth factor 1 and 2 (VEGFR1, VEGFR2), von Willebrand factor (vWF), Neuropillin (NRP1) and Endoglin (ENG) (Fig. 1g and S1c). To test the functionality of WT and FOP iECs in vitro, we show the formation of vessel like networks in a tube formation assay (Fig. S1d) and validate the formation of a tight endothelial barrier by impedance measurements, depicted as constant resistance values (Fig. 1f). Treatment with VEGFA caused decreased resistance with a subsequent recovery in iECs, similar to HUVECs (Fig. 1f). Moreover, endothelial response to pro-inflammatory TNF-α with a pro-adhesive phenotype was confirmed by expression of intracellular adhesion molecule-1 (ICAM-1) in iECs (Fig. 1h). In summary, WT and FOP iPSCs followed distinct ActivinA/SMAD responses and differentiated to ECs with high efficiencies without differences in the here assessed EC characteristics.

Generation of FOP iECs from iPSCs. (a) Representative Western blot of iPSCs after stimulation with different doses of ActivinA, BMP6 for 30 min (b) Workflow of iEC differentiation [21] with modification in red, created with BioRender.com. Below, phase contrast images of cells at different stages. (c) iEC differentation efficiency by FACS of CD144+ cells (WT-1; FOP1) at day 6. (d) Phase contrast and immunofluorescence stainings of EC markers CD144 and CD31 (iEC WT-1, FOP-1, HUVEC). (E) Representative FACS of CD144 and CD31 (WT-1, FOP-1, HUVEC). (F) Impedance measurement of VEGFA (2 nM) induced permeability of iEC and HUVEC monolayers. (G) RT-PCR of EC markers in iPSCs compared to iECs. Data is represented as mean normalized expression (MNE) ± SD, ****p < 0.0001 using one-way ANOVA (H) RT-PCR of ICAM1 expression upon 2 h TNFα (0.6 nM) treatment in iECs. Data is shown as mean fold induction (F.I.) ± SD ** p < 0.01 using two-way ANOVA
FOP iECs Show a SMAD1/5 Response to ActivinA
Interestingly, ACVR1 and ACVRL1 where the only BMP/TGFβ receptors upregulated during EC differentiation of iPSCs with no differences between WT and FOP (Fig. 2a and S2a). ActivinA (INHBA) expression was highly increased in WT and FOP iECs compared to iPSCs (Fig. 2b). Transcriptomics revealed that among the Activin/BMP ligands, INHBA and BMP6 showed the highest expression in iECs (Fig. S2b). While ActivinA increased pSMAD2 levels in WT and FOP iECs, strong SMAD1/5 phosphorylation was only seen in FOP iECs (Fig. 2c and S2c). ActivinA increased pSMAD1/5 levels dose- and time-dependently (Fig. 2c, Fig. S2c and S2d). In contrast, BMP6 induced phosphorylation of SMAD1/5 in WT and FOP iECs in a similar dose-dependent manner with slightly higher sensitivity of FOP iECs at 3 nM and 10 nM BMP6, indicating hypersensitive signaling (Fig. 2c and S2c).

Only FOP iECs gain SMAD1/5 responsiveness to ActivinA. (a) RT-PCR of type I receptors in iPSCs compared to iECs. Data is shown as MNE ± SD. (b) RT-PCR of INHBA in iPSCs compared to iECs. Data is shown as MNE ± SD. (c) Representative Western blot of iECs after stimulation with different doses of ActivinA, BMP6 for 30 min. (d) RT-PCR of BMP target genes upon 2 h BMP6 (5 nM), ActivinA (5 nM) treatment in iECs. Data is shown as mean F.I. ± SD. (E-F) Representative Western blot of iECs pretreated with type I receptor inhibitors (e) K02288 (0.5 μM) and treatment with BMP6, ActivinA for 30 min. (f) SB431542 (dose series) and treatment with ActivinA for 30 min. (MNE; mean normalized expression), (F.I.; fold induction), * p < 0.05,** p < 0.01, ***p < 0.001, ****p < 0.0001 significance was calculated using one-way (A,B) and two-way ANOVA (D, relative to unstimulated (w/o))
Aberrant ActivinA signaling was confirmed on SMAD1/5 target gene expression (ID1/2/3) in FOP iECs only, while BMP6 induced expression of ID1/2/3 in WT and FOP iECs (Fig. 2d and S2e). To verify the contribution of type I receptors to ActivinA/SMAD1/5 signaling, we pretreated iECs with K02288, a specific ACVR1/ACVRL1 inhibitor (Fig. 2e) or SB431542 (ACVR1B/C/TGFBR1 inhibitor) (Fig. 2f) and show prevention of SMAD1/5 phosphorylation only by K02288, confirming ActivinA signaling through ACVR1.
RNA-Seq Analysis Reveals a FOP-Specific Transcriptome upon ActivinA Stimulation
We performed whole transcriptome analysis of ActivinA treated WT and FOP iECs via RNA sequencing (RNASeq). Differentially expressed genes (DEG) were analyzed and compared between untreated and ActivinA (2 h) treated iECs from experimental replicates of each donor (Fig. 3a). Two independent FOP donors were stimulated with ActivinA and shared 212 DEG, whereof 64 showed a fold change (FC) of ≥1.5. Those genes were subjected to hierarchical cluster analysis comparing WT and FOP (Fig. 3c). The z-score indicates that most genes in FOP iECs were up- and only few were downregulated by ActivinA (Cluster a, b). In WT iECs, cluster a and b did not show any significant regulation upon ActivinA treatment except of sub-cluster b1, which included the SMAD2/3 target genes PMEPA1/TMEPAI and NEDD9 (Fig. 3c and S3a). Cluster b2 instead included SMAD1/5 target genes (e.g. ID1, ID3, SMAD6) (Fig. 3c). This indicates that ActivinA signaling leads to classical SMAD2/3 target gene transcription in WT and FOP iECs, whereas in FOP iECs additional genes were upregulated, including classical BMP/SMAD1/5 target genes. Accordingly, functional gene ontology (GO) annotation revealed significant association between upregulated genes and the BMP pathway and interestingly also the NOTCH pathway only in ActivinA treated FOP iECs (Fig. 3d). Integration of BMP and NOTCH signaling regulate vascular patterning of sprouting blood vessels [32], confirmed here as the GO analysis revealed blood vessel and vascular development in ActivinA treated FOP iECs (Fig. 3d and S3b). GO analysis of upregulated WT genes (FC of ≥1.5) identified TGF-β as the main associated signaling pathway and cell communication as the main biological function (p ≤ 0.05) (Fig. 3d and S3d). In summary, ActivinA induced pSMAD1/5 only in FOP iECs resulting in a FOP transcriptome consisting of highly enriched genes (e.g. ID1, NOG, HEY2, LFNG, UNC5B (Fig. 3e and S3c)), which are involved in blood vessel formation and activation of BMP and NOTCH pathways.

ActivinA induces a FOP-specific transcriptome in iECs. (a) Experimental setup: RNA Seq of 4 iEC lines, starved for 4 h and stimulated for 2 h with 5 nM ActivinA. (b) Venn diagram of RNASeq data presenting the number of DEG in both iEC donors (FOP-1 and FOP-2) stimulated with ActivinA. (c) Hierarchical clustering of shared DEG in both FOP donors (adjusted p value<0.05; −0.58 ≤ log2FC ≥0.58) of ActivinA treated and untreated (w/o) iECs. Heatmap color coding shows z-score of DEG (red = high; blue = low). Labeling „up”, „down “refers to DEG in FOP iECs upon ActivinA treatment. (d) Selected GO terms of shared upregulated genes in ActivinA treated FOP iECs and respective WT values. Depiction of log2 p value with Benjamini correction. Value is 0 if GO term was not identified. (e) Volcano Plot of DEG of ActivinA treated FOP-1 iECs. Genes (adjusted p value<0.05; −0.58 ≤ log2FC ≥0.58), up−/downregulation is indicated by color. Genes associated with GO terms are labeled. (DEG; Differentially expressed genes)
ActivinA and BMP6 Upregulate the Same Target Genes in FOP iECs
To further dissect whether ActivinA mediates the same downstream responses as BMP6, we treated cells with BMP6 at the same dose as ActivinA. While BMP6 upregulated the same BMP target genes (SMAD6, NOG, SMAD9, ID1/2/3) in WT and FOP iECs, ActivinA led to upregulation of those genes only in FOP (Fig. 4a, 2d and S2e). The same was observed for shared NOTCH target genes and those related to blood vessel formation (Fig. 4a), pointing towards an ActivinA specific mechanism in FOP, absent in WT iECs. Synergistic effects on NOTCH target genes by BMPs in regulating EC specification were reported previously [32]. Several NOTCH target genes, including LFNG, JAG1, HEY2, were confirmed as SMAD1/5 targets by chromatin immune precipitation sequencing [33]. Moreover, UNC5B and SGK1 were identified as EC-specific SMAD1/5 targets [33, 34]. However, to our knowledge, this is the first report interlinking ActivinA signaling to NOTCH target gene activation in ECs in the context of FOP.

Saracatinib rescues ActivinA/SMAD1/5 signaling responses in FOP iECs (a) RT-PCR validation of RNASeq target groups upon 1 h pretreatment with Saracatinib (0.2 μM) and 2 h ActivinA (5 nM), BMP6 (5 nM) treatment in iECs. Data is represented as mean F.I. ± SD. (b) Experimental setup: RNA Seq of 4 iEC lines, starved for 4 h, pretreated with Saracatinib and stimulated for 2 h with 5 nM ActivinA. (c) Independent hierarchical clustering of upregulated genes (adjusted p value<0.05; log2FC ≥0.58) in FOP iECs in comparison to WT and Saracatinib pretreated FOP iECs upon ActivinA (5 nM) treatment. Heatmap color coding shows z-score (red = high; blue = low). (d) Representative Western blot of of iECs pretreated with Saracatinib (0.2 μM) and ActivinA (5 nM) (F.I.; fold induction), * p < 0.05, ** p < 0.01, ***p < 0.001, ****p < 0.0001. Significance was calculated relative to unstimulated (w/o) using two-way ANOVA
Drug Candidate Saracatinib Rescues ActivinA/SMAD1/5 Signaling Responses in FOP iECs
The hypothesis that aberrant and hyperactivated SMAD1/5-dependent signaling of FOP- ACVR1 triggers HO in FOP has advanced the development of several drugs. Here, we used the kinase inhibitor Saracatinib (AZD-0530), a drug candidate for FOP [35] to investigate its inhibitory action in our in vitro disease model.
Saracatinib, initially discovered as a tyrosine kinase inhibitor and developed for the treatment of cancer [36] was later extended as an inhibitor for BMP type I receptors [37] and HO [38, 39]. The effect of Saracatinib on FOP endothelium has not been investigated yet. Here, we focused on early mechanistic actions of Saracatinib on endogenous ACVR1 signaling and its effect on the FOP transcriptome. iECs pretreated with Saracatinib for 1 h (Fig. 4b) followed by ActivinA stimulation inhibited pSMAD1/5 in both FOP donors, whereas pSMAD2 levels remained unaffected (Fig. 4d and S4a). Moreover, independent hierarchical cluster analysis of RNASeq data demonstrated rescue of the transcriptome induced by ActivinA in FOP iECs (Fig. 4c cluster a) to WT level after Saracatinib treatment (Fig. 4c cluster b1 and b2). Of note, Saracatinib preserved ActivinA induced transcription of the SMAD2/3 target genes in WT and FOP iECs (Fig. S3a and 4c cluster II).
Discussion
Early HO lesions are highly vascularized and FOP patient biopsies uncovered increased vessel number compared to non-hereditary HO [8]. Here, we optimized iPSC differentiation and present the first FOP iEC model, which recapitulates the gain of pathogenic ActivinA/SMAD1/5 signaling.
This is supported by a recent study showing ActivinA/SMAD1/5 signaling in primary ECs isolated from FOP patients [40] indicating that our iECs resemble primary ECs characteristics, providing a valuable, readily available source.
Our results differ from previous iPSC studies, which showed reduced viability [14] and no ActivinA/SMAD1/5 signaling in FOP iECs [15]. We found that FOP iPSCs already show aberrant ActivinA/SMAD1/5 signaling, prompting us to perform mesoderm induction only with exogenous BMP4 (without ActivinA supplementation) [21], while above mentioned studies used established methods for iEC generation (with exogenous ActivinA, BMP4) [41, 42]. BMP4 is essential for mesoderm formation in vivo [43] and in vitro [44], whereas ActivinA was shown to promote mesoderm in vitro but still relies on co-treated BMP4 [45]. Interestingly, different ActivinA, BMP4 doses give rise to multiple mesoderm subsets prior to EC formation in vitro [46], which may be an indication for different EC type generation. Advances in characterization of iEC types, differentiated via distinct routes will advance the understanding of EC heterogeneity in healthy and diseased human tissue.
The underlying mechanism of ActivinA/SMAD1/5 signaling is not fully understood but suggests ACVR1 dependency and independency of ACVR1B/C/TGFBR1 as demonstrated by inhibitor experiments in FOP iECs being in line with knockdown studies in FOP [12] and myeloma cells [47]. Downstream effects of ActivinA/SMAD1/5 signaling in human tissue remain poorly understood. Our study provides first insight of whole genome responses to ActivinA in ECs with upregulation of SMAD2/3 target genes in both WT and FOP iECs and upregulation of additional genes only in FOP iECs. This supports the model that WT-ACVR1 forms a non-signaling complex with ActivinA while binding of ActivinA to FOP-ACVR1 results in an active receptor complex promoting SMAD1/5 signaling [48].
In FOP iECs ActivinA upregulated classical BMP targets like Noggin, a negative feedback regulator that antagonizes certain BMPs but not ActivinA. Additionally, we show ActivinA induced upregulation of NOTCH target genes in FOP iECs. EC-specific disruption of NOTCH signaling in mice impaired bone vessel growth and reduced osteogenesis [49], suggesting that ActivinA could promote coupled angiogenesis and osteogenesis in FOP lesions via NOTCH activation. BMPs synergize or antagonize with NOTCH on different levels to control tip and stalk cell shuffling in blood vessel branching and vascular patterning [1, 32]. We confirmed that ActivinA upregulated the same genes as BMP6 in FOP iECs indicating that mutant ACVR1 lacks ligand specificity by transducing a BMP-like response.
Notably, a model of dynamic pSMAD1/5 regulation in blood vessel development suggests that pSMAD1/5 activates distinct target genes in single ECs thereby pre-patterning the endothelium for tip/stalk cell mediated sprouting [50]. Interestingly, in our study, genes associated with stalk cell identity such as JAG1, HEY2 were upregulated only in FOP iECs by ActivinA. This is in line with BMP6 induced stalk cell genes via ACVR1 signaling in HUVECs [51].
Therefore, we propose that the ActivinA induced transcriptome via ACVR1/SMAD1/5 signaling pre-patterns the FOP endothelium, which affects tip/stalk cell shuffling in new formed blood vessels in early HO lesions. Potentially, this underlying molecular mechanism contributes to the vascular phenotype found in HO lesions of FOP patients.
However, to functionally recapitulate the complex vascular phenotype in FOP lesions during HO we suggest to integrate iECs in skeletal muscle organoid models [52] to establish a human FOP organoid for future studies. Very recently, fibroblasts were identified as the main source of ActivinA during HO of FOP mice [53]. Fibroblasts proliferate in early pre-osseous HO lesions accompanied by neovascularization [5]. Thus, our iECs can also be used to model human vascularized, fibroproliferative lesions in co-culture experiments with fibroblasts.
Drug testing was performed to rescue the ActivinA induced transcriptome in FOP iECs. The drug candidate Saracatinib prevented only aberrant pSMAD1/5 by ActivinA in FOP iECs, remained SMAD2/3 signaling and successfully restored the FOP transcriptome to WT expression levels. This suggests prevention of aberrant ActivinA effects on the vasculature during HO in FOP patients by the drug candidate Saracatinib and contributes to a better understanding of the specific mechanistic action of Saracatinib in human tissues. Very recently, first clinical investigations of Saracatinib in FOP were initiated (NCT04307953). Thus, we have established iECs as a powerful patient model for further studies on disease mechanism(s) under endogenous receptor levels and for drug testing.
References
Jin, Y., Kaluza, D., & Jakobsson, L. (2014). VEGF, notch and TGFβ/BMPs in regulation of sprouting angiogenesis and vascular patterning. Biochemical Society Transactions, 42(6), 1576–1583. https://doi.org/10.1042/BST20140231.
Chung, A. S., Lee, J., & Ferrara, N. (2010). Targeting the tumour vasculature: Insights from physiological angiogenesis. Nature Reviews Cancer, 10(7), 505–514. https://doi.org/10.1038/nrc2868.
Meyers, C., Lisiecki, J., Miller, S., Levin, A., Fayad, L., Ding, C., Sono, T., McCarthy, E., Levi, B., & James, A. W. (2019). Heterotopic ossification: A comprehensive review. JBMR plus, 3(4), e10172. https://doi.org/10.1002/jbm4.10172.
Cocks, M., Mohan, A., Meyers, C. A., Ding, C., Levi, B., McCarthy, E., & James, A. W. (2017). Vascular patterning in human heterotopic ossification. Human Pathology, 63, 165–170. https://doi.org/10.1016/j.humpath.2017.03.005.
Shore, E. M., & Kaplan, F. S. (2010). Inherited human diseases of heterotopic bone formation. Nature Reviews Rheumatology, 6(9), 518–527. https://doi.org/10.1038/nrrheum.2010.122.
Kaplan, F. S., Tabas, J. A., Gannon, F. H., Finkel, G., Hahn, G. V., & Zasloff, M. A. (1993). The histopathology of fibrodysplasia ossificans progressiva. An endochondral process. The Journal of Bone and Joint Surgery. American Volume, 75(2), 220–230. https://doi.org/10.2106/00004623-199302000-00009.
Hwang, C., Marini, S., Huber, A. K., Stepien, D. M., Sorkin, M., Loder, S., Pagani, C. A., Li, J., Visser, N. D., Vasquez, K., Garada, M. A., Li, S., Xu, J., Hsu, C. Y., Yu, P. B., James, A. W., Mishina, Y., Agarwal, S., Li, J., & Levi, B. (2019). Mesenchymal VEGFA induces aberrant differentiation in heterotopic ossification. Bone Research, 7(1), 1–17. https://doi.org/10.1038/s41413-019-0075-6.
Ware, A. D., Brewer, N., Meyers, C., Morris, C., McCarthy, E., Shore, E. M., & James, A. W. (2019). Differential vascularity in genetic and nonhereditary heterotopic ossification. International Journal of Surgical Pathology, 27(8), 859–867. https://doi.org/10.1177/1066896919857135.
Shore, E. M., Xu, M., Feldman, G. J., Fenstermacher, D. A., Cho, T. J., Choi, I. H., Connor, J. M., Delai, P., Glaser, D. L., LeMerrer, M., Morhart, R., Rogers, J. G., Smith, R., Triffitt, J. T., Urtizberea, J. A., Zasloff, M., Brown, M. A., & Kaplan, F. S. (2006). A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature Genetics, 38(5), 525–527. https://doi.org/10.1038/ng1783.
Shen, Q., Little, S. C., Xu, M., Haupt, J., Ast, C., Katagiri, T., Mundlos, S., Seemann, P., Kaplan, F. S., Mullins, M. C., & Shore, E. M. (2009). The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. The Journal of Clinical Investigation, 119(11), 3462–3472. https://doi.org/10.1172/JCI37412.
Hatsell, S. J., Idone, V., Wolken, D. M. A., Huang, L., Kim, H. J., Wang, L., et al. (2015). ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin a. Science Translational Medicine, 7(303), 303ra137. https://doi.org/10.1126/scitranslmed.aac4358.
Hino, K., Ikeya, M., Horigome, K., Matsumoto, Y., Ebise, H., Nishio, M., Sekiguchi, K., Shibata, M., Nagata, S., Matsuda, S., & Toguchida, J. (2015). Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proceedings of the National Academy of Sciences of the United States of America, 112(50), 15438–15443. https://doi.org/10.1073/pnas.1510540112.
Yadin, D., Knaus, P., & Mueller, T. D. (2016). Structural insights into BMP receptors: Specificity, activation and inhibition. Cytokine & Growth Factor Reviews, 27, 13–34. https://doi.org/10.1016/j.cytogfr.2015.11.005.
Cai, J., Orlova, V. V., Cai, X., Eekhoff, E. M. W., Zhang, K., Pei, D., Pan, G., Mummery, C. L., & ten Dijke, P. (2015). Induced pluripotent stem cells to model human Fibrodysplasia Ossificans Progressiva. Stem Cell Reports, 5(6), 963–970. https://doi.org/10.1016/j.stemcr.2015.10.020.
Barruet, E., Morales, B. M., Lwin, W., White, M. P., Theodoris, C. V., Kim, H., Urrutia, A., Wong, S. A., Srivastava, D., & Hsiao, E. C. (2016). The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling. Stem Cell Research & Therapy, 7(1), 115. https://doi.org/10.1186/s13287-016-0372-6.
Hildebrand, L., Rossbach, B., Kühnen, P., Gossen, M., Kurtz, A., Reinke, P., Seemann, P., & Stachelscheid, H. (2016). Generation of integration free induced pluripotent stem cells from fibrodysplasia ossificans progressiva (FOP) patients from urine samples. Stem Cell Research, 16(1), 54–58. https://doi.org/10.1016/j.scr.2015.11.017.
Rossbach, B., Hildebrand, L., El-Ahmad, L., Stachelscheid, H., Reinke, P., & Kurtz, A. (2016). Generation of a human induced pluripotent stem cell line from urinary cells of a healthy donor using an integration free vector. Stem Cell Research, 16(2), 314–317. https://doi.org/10.1016/j.scr.2015.12.018.
Rossbach, B., Hildebrand, L., El-Ahmad, L., Stachelscheid, H., Reinke, P., & Kurtz, A. (2017). Generation of a human induced pluripotent stem cell line from urinary cells of a healthy donor using integration free Sendai virus technology. Stem Cell Research, 21, 167–170. https://doi.org/10.1016/j.scr.2016.09.002.
Chen, G., Gulbranson, D. R., Hou, Z., Bolin, J. M., Ruotti, V., Probasco, M. D., Smuga-Otto, K., Howden, S. E., Diol, N. R., Propson, N. E., Wagner, R., Lee, G. O., Antosiewicz-Bourget, J., Teng, J. M. C., & Thomson, J. A. (2011). Chemically defined conditions for human iPSC derivation and culture. Nature Methods, 8(5), 424–429. https://doi.org/10.1038/nmeth.1593.
Wong, K. G., Ryan, S. D., Ramnarine, K., Rosen, S. A., Mann, S. E., Kulick, A., de Stanchina, E., Müller, F. J., Kacmarczyk, T. J., Zhang, C., Betel, D., & Tomishima, M. J. (2017). CryoPause: A new method to immediately initiate experiments after cryopreservation of pluripotent stem cells. Stem Cell Reports, 9(1), 355–365. https://doi.org/10.1016/j.stemcr.2017.05.010.
Patsch, C., Challet-Meylan, L., Thoma, E. C., Urich, E., Heckel, T., O’Sullivan, J. F., Grainger, S. J., Kapp, F. G., Sun, L., Christensen, K., Xia, Y., Florido, M. H. C., He, W., Pan, W., Prummer, M., Warren, C. R., Jakob-Roetne, R., Certa, U., Jagasia, R., Freskgård, P. O., Adatto, I., Kling, D., Huang, P., Zon, L. I., Chaikof, E. L., Gerszten, R. E., Graf, M., Iacone, R., & Cowan, C. A. (2015). Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nature Cell Biology, 17(8), 994–1003. https://doi.org/10.1038/ncb3205.
Ren, X., Moser, P. T., Gilpin, S. E., Okamoto, T., Wu, T., Tapias, L. F., Mercier, F. E., Xiong, L., Ghawi, R., Scadden, D. T., Mathisen, D. J., & Ott, H. C. (2015). Engineering pulmonary vasculature in decellularized rat and human lungs. Nature Biotechnology, 33(10), 1097–1102. https://doi.org/10.1038/nbt.3354.
Pfaffl, M. W. (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research, 29(9), e45. https://doi.org/10.1093/nar/29.9.e45.
Afgan, E., Baker, D., Batut, B., van den Beek, M., Bouvier, D., Čech, M., Chilton, J., Clements, D., Coraor, N., Grüning, B. A., Guerler, A., Hillman-Jackson, J., Hiltemann, S., Jalili, V., Rasche, H., Soranzo, N., Goecks, J., Taylor, J., Nekrutenko, A., & Blankenberg, D. (2018). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Research, 46(W1), W537–W544. https://doi.org/10.1093/nar/gky379.
Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., & Gingeras, T. R. (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15–21. https://doi.org/10.1093/bioinformatics/bts635.
Ewels, P., Magnusson, M., Lundin, S., & Käller, M. (2016). MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics, 32(19), 3047–3048. https://doi.org/10.1093/bioinformatics/btw354.
Kovaka, S., Zimin, A. V., Pertea, G. M., Razaghi, R., Salzberg, S. L., & Pertea, M. (2019). Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biology, 20(1), 278. https://doi.org/10.1186/s13059-019-1910-1.
Love, M. I., Huber, W., & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 550. https://doi.org/10.1186/s13059-014-0550-8.
Hulsen, T., de Vlieg, J., & Alkema, W. (2008). BioVenn – A web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics, 9(1), 488. https://doi.org/10.1186/1471-2164-9-488.
Huang, D. W., Sherman, B. T., & Lempicki, R. A. (2009). Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research, 37(1), 1–13. https://doi.org/10.1093/nar/gkn923.
Huang, D. W., Sherman, B. T., & Lempicki, R. A. (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols, 4(1), 44–57. https://doi.org/10.1038/nprot.2008.211.
Beets, K., Huylebroeck, D., Moya, I. M., Umans, L., & Zwijsen, A. (2013). Robustness in angiogenesis: Notch and BMP shaping waves. Trends in genetics: TIG, 29(3), 140–149. https://doi.org/10.1016/j.tig.2012.11.008.
Morikawa, M., Koinuma, D., Tsutsumi, S., Vasilaki, E., Kanki, Y., Heldin, C.-H., Aburatani, H., & Miyazono, K. (2011). ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Research, 39(20), 8712–8727. https://doi.org/10.1093/nar/gkr572.
Araki, M., Hisamitsu, T., Kinugasa-Katayama, Y., Tanaka, T., Harada, Y., Nakao, S., Hanada, S., Ishii, S., Fujita, M., Kawamura, T., Saito, Y., Nishiyama, K., Watanabe, Y., & Nakagawa, O. (2018). Serum/glucocorticoid-regulated kinase 1 as a novel transcriptional target of bone morphogenetic protein-ALK1 receptor signaling in vascular endothelial cells. Angiogenesis, 21(2), 415–423. https://doi.org/10.1007/s10456-018-9605-x.
Kitoh, H. (2020). Clinical aspects and current therapeutic approaches for FOP. Biomedicines, 8(9), 325. https://doi.org/10.3390/biomedicines8090325.
Hennequin, L. F., Allen, J., Breed, J., Curwen, J., Fennell, M., Green, T. P., Lambert-van der Brempt, C., Morgentin, R., Norman, R. A., Olivier, A., Otterbein, L., Plé, P. A., Warin, N., & Costello, G. (2006). N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5- (tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. Journal of Medicinal Chemistry, 49(22), 6465–6488. https://doi.org/10.1021/jm060434q.
Lewis, T. C., & Prywes, R. (2013). Serum regulation of Id1 expression by a BMP pathway and BMP responsive element. Biochimica et Biophysica Acta, 1829(10), 1147–1159. https://doi.org/10.1016/j.bbagrm.2013.08.002.
Wentworth, K. L., Masharani, U., & Hsiao, E. C. (2019). Therapeutic advances for blocking heterotopic ossification in fibrodysplasia ossificans progressiva. British Journal of Clinical Pharmacology, 85(6), 1180–1187. https://doi.org/10.1111/bcp.13823.
Hino, K., Zhao, C., Horigome, K., Nishio, M., Okanishi, Y., Nagata, S., Komura, S., Yamada, Y., Toguchida, J., Ohta, A., & Ikeya, M. (2018). An mTOR signaling modulator suppressed heterotopic ossification of Fibrodysplasia Ossificans Progressiva. Stem Cell Reports, 11(5), 1106–1119. https://doi.org/10.1016/j.stemcr.2018.10.007.
Sánchez-Duffhues, G., Williams, E., Benderitter, P., Orlova, V., van Wijhe, M., Garcia de Vinuesa, A., et al. (2019). Development of macrocycle kinase inhibitors for ALK2 using Fibrodysplasia Ossificans Progressiva-derived endothelial cells. JBMR Plus, 3(11), e10230. https://doi.org/10.1002/jbm4.10230.
Orlova, V. V., van den Hil, F. E., Petrus-Reurer, S., Drabsch, Y., Ten Dijke, P., & Mummery, C. L. (2014). Generation, expansion and functional analysis of endothelial cells and pericytes derived from human pluripotent stem cells. Nature Protocols, 9(6), 1514–1531. https://doi.org/10.1038/nprot.2014.102.
White, M. P., Rufaihah, A. J., Liu, L., Ghebremariam, Y. T., Ivey, K. N., Cooke, J. P., & Srivastava, D. (2013). Limited gene expression variation in human embryonic stem cell and induced pluripotent stem cell-derived endothelial cells. Stem Cells (Dayton, Ohio), 31(1), 92–103. https://doi.org/10.1002/stem.1267.
Winnier, G., Blessing, M., Labosky, P. A., & Hogan, B. L. (1995). Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes & Development, 9(17), 2105–2116. https://doi.org/10.1101/gad.9.17.2105.
Zhang, P., Li, J., Tan, Z., Wang, C., Liu, T., Chen, L., Yong, J., Jiang, W., Sun, X., du, L., Ding, M., & Deng, H. (2008). Short-term BMP-4 treatment initiates mesoderm induction in human embryonic stem cells. Blood, 111(4), 1933–1941. https://doi.org/10.1182/blood-2007-02-074120.
Cerdan, C., McIntyre, B. A. S., Mechael, R., Levadoux-Martin, M., Yang, J., Lee, J. B., & Bhatia, M. (2012). Activin a promotes hematopoietic fated mesoderm development through Upregulation of Brachyury in human embryonic stem cells. Stem Cells and Development, 21(15), 2866–2877. https://doi.org/10.1089/scd.2012.0053.
Era, T., Izumi, N., Hayashi, M., Tada, S., Nishikawa, S., & Nishikawa, S.-I. (2008). Multiple mesoderm subsets give rise to endothelial cells, whereas hematopoietic cells are differentiated only from a restricted subset in embryonic stem cell differentiation culture. Stem Cells, 26(2), 401–411. https://doi.org/10.1634/stemcells.2006-0809.
Olsen, O. E., Hella, H., Elsaadi, S., Jacobi, C., Martinez-Hackert, E., & Holien, T. (2020). Activins as dual specificity TGF-β family molecules: SMAD-activation via Activin- and BMP-type 1 receptors. Biomolecules, 10(4), 519. https://doi.org/10.3390/biom10040519.
Aykul, S., Corpina, R, A., Goebel, E, J., Cunanan, C, J., Dimitriou, A., Kim, H., … Idone, V. (2020). Activin a forms a non-signaling complex with ACVR1 and type II Activin/BMP receptors via its finger 2 tip loop. eLife. eLife Sciences Publications Limited. https://doi.org/10.7554/eLife.54582, 9.
Ramasamy, S. K., Kusumbe, A. P., Wang, L., & Adams, R. H. (2014). Endothelial notch activity promotes angiogenesis and osteogenesis in bone. Nature, 507(7492), 376–380. https://doi.org/10.1038/nature13146.
Moya, I. M., Umans, L., Maas, E., Pereira, P. N. G., Beets, K., Francis, A., Sents, W., Robertson, E. J., Mummery, C. L., Huylebroeck, D., & Zwijsen, A. (2012). Stalk cell phenotype depends on integration of notch and Smad1/5 signaling cascades. Developmental Cell, 22(3), 501–514. https://doi.org/10.1016/j.devcel.2012.01.007.
Benn, A., Hiepen, C., Osterland, M., Schutte, C., Zwijsen, A., & Knaus, P. (2017). Role of bone morphogenetic proteins in sprouting angiogenesis: Differential BMP receptor-dependent signaling pathways balance stalk vs. tip cell competence. The FASEB Journal, 31(11), 4720–4733. https://doi.org/10.1096/fj.201700193RR.
Maffioletti, S. M., Sarcar, S., Henderson, A. B. H., Mannhardt, I., Pinton, L., Moyle, L. A., Steele-Stallard, H., Cappellari, O., Wells, K. E., Ferrari, G., Mitchell, J. S., Tyzack, G. E., Kotiadis, V. N., Khedr, M., Ragazzi, M., Wang, W., Duchen, M. R., Patani, R., Zammit, P. S., Wells, D. J., Eschenhagen, T., & Tedesco, F. S. (2018). Three-dimensional human iPSC-derived artificial skeletal muscles model muscular dystrophies and enable multilineage tissue engineering. Cell Reports, 23(3), 899–908. https://doi.org/10.1016/j.celrep.2018.03.091.
Hwang, C., Pagani, C. A., Das, N., Marini, S., Huber, A. K., Xie, L., Jimenez, J., Brydges, S., Lim, W. K., Nannuru, K. C., Murphy, A. J., Economides, A. N., Hatsell, S. J., & Levi, B. (2020). Activin a does not drive post-traumatic heterotopic ossification. Bone, 115473, 115473. https://doi.org/10.1016/j.bone.2020.115473.
Acknowledgements
We thank Petra Seemann, Bella Rossbach, Andreas Kurtz for kindly providing the iPSC lines. We thank Iris Fischer, Tanja Fisch and the BIH Stem Cell Core Facility team for technical assistance and feedback. We acknowledge the support of FOP Germany e.V.. For critical feedback and advice we thank Nadine Großmann, Jerome Jatzlau, Paul Mendez and all members of the Knaus lab. The graphical abstract was created with BioRender.com.
Funding
Open Access funding enabled and organized by Projekt DEAL. The authors acknowledge support from Deutsche Forschungsgemeinschaft DFG: SFB 958, FOR 2165 and BSRT; Einstein Center for Regenerative Therapies and FOP Germany e.V..
Author information
Authors and Affiliations
Contributions
Writing-original draft preparation: S.H.; Writing-review and editing: S.H., P.K., J.H., H.S., L.H.; Conceptualization: P.K., S.H.; Investigation: S.H., B.K., K.F.; Methodology: S.H., K.F., L.H.; Formal analysis: S.H.; Resources: P.K., H.S., L.H.; Funding Acquisition: P.K., H.S., S.H; Supervision: P.K., H.S., J.H., S.H.
Corresponding author
Ethics declarations
Declaration of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Figure S1
iPSC and iEC characterization. Related to Fig. 1. (a) Sequencing of genomic DNA of iECs from 4 donors at locus of ACVR1 R206H mutation. Arrows indicate point mutation. (b) Representative Western blot of lysates from iPSC after stimulation with different doses of ActivinA, BMP6 for 30 min. (c) RT-PCR of EC marker in iPSCs compared to iECs. Data is shown as mean normalized expression (MNE) ± SD. * p < 0.05, *** p < 0.001,****p < 0.0001 using one-way ANOVA. (d) Representative phase contrast images of tube-like structures formed by WT and FOP iEs on Matrigel after 24 h. (PNG 2079 kb)
Figure S2
ACVR1 signaling characteristics in iECs. Related to Fig. 2. (a) RT-PCR of type II receptors in iPSCs compared to iECs. Data is shown as MNE ± SD. (b) Relative expression of the TGFβ ligand family shown as FPKM values of RNASeq data of untreated iECs. (c) Representative Western blot of lysates from iECs after stimulation with different doses of ActivinA, BMP6 for 30 min. (d) or with ActivinA (5 nM) for different time points. (e) RT-PCR of BMP target gene ID3 upon 2 h BMP6 (5 nM), ActivinA (5 nM) treatment in iECs. Data is shown as mean F.I. ± SD. (f) (F.I.; fold induction), * p < 0.05,**,** p < 0.01,****p < 0.0001. Significance was calculated using one-way (A) and two-way ANOVA (E, relative to unstimulated (w/o)). (PNG 1329 kb)
Figure S3
ActivinA downstream signaling responses in iECs. Related to Fig. 3. (a) RT-PCR of SMAD2/3 target genes upon (1 h pretreatment with Saracatinib 0.2 μM) and 2 h ActivinA (5 nM) in iECs. Data is shown as mean F.I. ± SD. (b) GO terms of upregulated genes in ActivinA treated FOP iECs. Depiction of log2 p value of Benjamini correction (cut-off at adjusted p value<0.01). (c) Volcano Plot of differentially expressed genes of ActivinA treated FOP-2 iECs. Genes of (adjusted p value<0.05; −0.58 ≤ log2FC ≥0.58) up−/downregulation is indicated by color. Genes associated with GO terms are labeled. (d) GO terms of upregulated genes in ActivinA treated WT iECs. Depiction of all log2 p values of Benjamini correction (adjusted p value<0.05). (PNG 1607 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hildebrandt, S., Kampfrath, B., Fischer, K. et al. ActivinA Induced SMAD1/5 Signaling in an iPSC Derived EC Model of Fibrodysplasia Ossificans Progressiva (FOP) Can Be Rescued by the Drug Candidate Saracatinib. Stem Cell Rev and Rep 17, 1039–1052 (2021). https://doi.org/10.1007/s12015-020-10103-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12015-020-10103-9