
Abstract
Purpose of review
Pulmonary hypertension is characterized by an elevation of pulmonary artery pressures and prolonged exposure of the right ventricle to high afterload that collectively contribute to morbidity and mortality in both the term and preterm infants. This review summarizes the pathogenesis, etiologies, and hemodynamic profiles of the conditions that result in pulmonary hypertension in neonates. We explore the application of echocardiographic techniques for the assessment of right ventricular performance and pulmonary hemodynamics that enhance and guide the diagnosis and management strategies in neonates.
Recent findings
Clinical assessments based on the determinants of mean pulmonary artery pressures (pulmonary vascular resistance, pulmonary blood flow, and pulmonary capillary wedge pressure) provide a physiologic approach in determining the acute and chronic etiologies of pulmonary hypertension in neonates. In addition, advances in neonatal echocardiography now afford the capability to obtain quantitative information that often precedes the qualitative information acquired by conventional methods and also provide sensitive markers of right ventricle performance for prognostic information based on the determinants of mean pulmonary artery pressures.
Summary
Neonatal pulmonary hypertension represents a physiologic spectrum that accounts for the variance in clinical presentation and response to therapies. Physiology-based approaches to etiological identification, coupled with the emerging echocardiographic methods for the assessment of pulmonary hypertension in neonates will likely help to identify cardiovascular compromise earlier, guide therapeutic intervention, monitor therapeutic effectiveness, and improve overall outcome.
Similar content being viewed by others

Introduction
Pulmonary hypertension (PHT) is characterized by a state of sustained elevation of pulmonary artery pressures (PAP) and prolonged exposure of the right ventricle (RV) to high afterload that collectively contribute to morbidity and mortality in both the term and preterm infants. The diagnosis and management of the hemodynamic status of neonates with PHT are challenging, owing to the multitude of etiologies and the unique characteristics of the pulmonary circulatory system. Although mean PAP (mPAP) is directly related to pulmonary blood flow (PBF) and pulmonary capillary wedge pressure (PWCP), the pathophysiological hallmark of neonatal PHT is increased pulmonary vascular resistance (PVR) [1••]. In acute PHT (aPHT), failure of reduction in PVR during the postnatal transitional period results in impaired oxygenation, RV dysfunction, and pulmonary-to-systemic shunting. Chronic PHT (cPHT) may also result from exposure to high PBF or PCWP but traditionally occurs secondary to a rise in PVR beyond the first month of age (often with initial successful postnatal transition) that is seen most frequently with chronic neonatal lung diseases. In this review, we discuss the embryologic origins of RV and pulmonary circulatory development and the pathogenesis, various etiologies, and hemodynamic profiles of PHT in term and preterm neonates. We examine the application of echocardiographic techniques that enhance and guide the diagnosis and management strategies in neonates.
Pulmonary circulation and right ventricle development
Cardiac morphogenesis initially precedes airway development, but distinctive elements of the pulmonary system arise from primitive heart tube during the embryonic stage [2]. The genetic investigation of cardiac morphogenesis has shown that the cells of the anterior second heart field develop into the RV during the looping process and form the main pulmonary arterial trunk [3]. The pulmonary arterial precursors then form a multilayered vascular network linking the arterial and venous poles of the heart with a continuous circulation between the RV and lungs. The pulmonary circulation subsequently arises shortly thereafter through temporal and spatially controlled signaling pathways linked to both the cardiac and airway development [3]. The RV and pulmonary circulation are separated from the left ventricle (LV) and systemic circulation by atrial and ventricular septation, although they remain a parallel circulation during fetal life through a patent foramen ovale and ductus arteriosus. At birth, inflation of the lungs, increased oxygen tension, and appropriate reduction in PVR allow increased pulmonary blood flow to the left side of the heart resulting in closure of the remaining fetal shunts.
The RV develops as a complex tripartite structure made up of an inflow area, trabeculated apex, and outlet infundibulum leading into the pulmonary arterial circulation. The coarse trabeculations and thin-walled structure allow the RV to acutely dilate to accommodate increases in volume and afterload. The RV is the dominant chamber in the fetal period, supplying 45–60% of the total cardiac output depending on the stage of gestation [4]. The pulmonary arterial circulation consists of thin, elastic vessels that accompany the arborization of the bronchial airway but remain constricted by vasoactive mediators. Only 15–25% of the total cardiac output circulates through the pulmonary vasculature with the remaining RV output redirected through the ductus arteriosus (DA) into the systemic circulation.
Etiopathogenesis of pulmonary hypertension
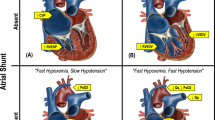
Mean PAP (mPAP) is directly proportional to PBF, PVR, and PCWP and can be summarized as the following equation: mPAP = (PBF × PVR) + PCWP [1••]. An increase in any of the three determinants of mPAP can lead to PHT (Fig. 1). Flow-related PHT has acutely been reported in the context of arterial venous malformations [5, 6] and chronically with left-to-right intra- and extra-cardiac shunts, e.g., atrial septal defect (ASD), ventricular septal defects (VSD), and patent DA (PDA) [1••, 7]. The hemodynamically significant left-to-right shunts can result in chronic pulmonary overcirculation adding an extra stressor to the immature pulmonary vasculature in the preterm infant and predisposing it to vascular remodeling and potential development of PHT. Increased PCWP may also lead to pulmonary venous congestion and PHT in neonates. Etiologies include left atrial and ventricular dysfunction seen in prematurity [8] and infants with hypoxic-ischemic encephalopathy (HIE) [9]. Pulmonary vein stenosis may also result in PHT in premature infants and has been observed with growing frequency in infants with chronic lung disease (CLD) [10]. Although the exact mechanistic link is unknown, possible pathways include abnormal angiogenesis related to CLD, chronic pulmonary overcirculation from concomitant left-to-right shunting lesions, and increased PCWP [10].

Etiopathology-based classification of neonatal pulmonary hypertension. AVM, arterial venous malformations; L-to-R, left to right; TAPVC, total anomalous pulmonary vein connection; RDS, respiratory distress syndrome; CLD, chronic lung disease; MAS, meconium aspiration syndrome; CDH, congenital diaphragmatic hernia; SSRI, selective serotonin receptor inhibitor; NSAID, nonsteroidal anti-inflammatory drug; VSD, ventricular septal defect; SPD, surfactant protein deficiency disorders; ABCA3, ATP binding cassette subfamily A member 3; HIE, hypoxic ischemic encephalopathy; AS, aortic stenosis; MS, mitral stenosis. Words in boldface indicate etiological entities of particular interest in prematurely born neonates.
Though the above mechanisms of elevated PBF and PCWP lead to raised pulmonary pressures, in practice, increased PVR resulting from a combination of intrinsic, secondary, and acquired abnormalities in the pulmonary vasculature is widely recognized as the pathophysiologic hallmark of neonatal PHT. In the absence of intracardiac shunts, PVR is defined as the transpulmonary pressure gradient divided by PBF and refers to the resistance that the RV must overcome to provide antegrade flow to the pulmonary circulation. The major determinants of PVR are the pulmonary arterial count, capillary vascular bed structure, and vasomotor tone. There are five cellular pathways involved in regulation of pulmonary vascular tone: (1) nitric oxide (NO)–soluble guanylate cyclase–cyclic guanylyl monophosphate (cGMP); (2) prostaglandin–prostacyclin–cyclic adenosine monophosphate (cAMP); (3) RhoKinase; (4) endothelin; (5) reactive oxygen species (ROS) [7]. Isolated abnormal vasoreactivity from interruption of these cellular pathways in combination with structurally abnormal pulmonary vasculature are the major causes of high PVR-related PHT in neonates.
In the fetal circulation, the highly vascular, low-resistance placenta serves as the organ for gas exchange, receiving oxygenated blood from the maternal circulation and contributing to a lowered fetal systemic vascular resistance (SVR) [11, 12]. The lungs only receive a small amount of blood flow due to the vasoconstricted state (high PVR) of the fetal pulmonary vasculature [12]. In utero pulmonary vasoconstriction is maintained by several mechanisms, including fetal lung fluid, low oxygen tension, release of the endogenous vasoconstrictors (e.g., endothelin-1, thromboxane, and platelet-activating factor), ROS, and increased Rho A-Rho Kinase [13]. The immediate postnatal period is regulated by the complex interplay between local vasoconstrictor and vasodilator mediators and transitional circulatory events to ensure a smooth move from fetal to extrauterine life. Clamping of the umbilical cord removes the low-resistance placental flow with a subsequent rise in the systemic afterload (SVR), and the first breath of oxygenated air results in lung expansion and relief of alveolar hypoxia that dramatically decreases PVR leaving the postnatal pulmonary circulation as a low pressure, low resistance, high flow system. Pulmonary vasodilation is further regulated by postnatal mechanical distention, decrease in carbon dioxide tension, increase in oxygen tension, surge of vasodilator prostaglandins, and release of NO [14, 15]. Conditions that interfere with the normal postnatal decline in the PVR/SVR ratio coupled with mediators that promote vasoconstriction and negate vasodilation cause the transitional circulation to persist and result in aPHT.
There are three types of etiological abnormalities of the pulmonary vasculature that underlie PHT and lead to elevated PVR. [1] Primary or idiopathic PHT is characterized by remodeling of the pulmonary vessels (structure and number) with vascular wall thickening and smooth muscle hyperplasia. The pulmonary vasculature is maldeveloped with normal lung parenchyma. As the smooth muscle extends to the level of the intra-acinar arteries, a process that normally occurs much later in the postnatal period, the pulmonary vasculature will not vasodilate appropriately in response to birth-related stimuli. In addition, there is disruption of the cellular signaling pathways and these neonates present with profound hypoxemia and clear hyperlucent lung fields. [2] In secondary PHT, the pulmonary vasculature is structurally normal but with abnormal vasoreactivity affecting the transitional reduction of PVR; this can often be reversed. The pulmonary vasculature maladapts at birth due to parenchymal lung disease processes (e.g., respiratory distress syndrome (RDS), meconium aspiration syndrome, pneumonia, and air leak) that affect oxygenation, ventilation, and lung recruitment. Extra-parenchymal disorders such as perinatal asphyxia with acidosis and sepsis can also contribute to this phenomenon [1••]. The third common cause of PVR-related PHT is pulmonary vascular hypoplasia that is underdeveloped and often refractory or irreversible, depending on the nature of underlying involvement [7]. Primary causes include alveolar capillary dysplasia with misalignment of pulmonary veins, pulmonary interstitial lymphangiectasia, primary surfactant metabolism disorders, or congenital diaphragmatic hernia (CDH). Secondary causes are associated with restrictive lung growth and absence of/decreased fetal breathing.
While this traditional etiologic categorization of pulmonary vasculature is helpful, a more recent approach complements the current classification by differentiating neonatal PHT by its clinical phenotype, aPHT or cPHT [1••]. aPHT presents in the immediate postnatal period secondary to abnormal transition of the pulmonary circulation from a high-resistance intrauterine to a low-resistance extrauterine circuit. This is often referred to as persistent pulmonary hypertension of the newborn (PPHN). However, the term “PPHN” can misrepresent the normal physiologic process in the transitional period, as elevated mPAP is ubiquitous [16••]. Giesinger et al. [16••] suggest the term “aPHT” may be more appropriate because it provides an accurate representation of the pathophysiologic disturbances with elevated mPAP, oxygenation failure, and potential RV dysfunction. In contrast, “chronic PHT” in neonates is secondary to a rise in PVR outside of the transition period and can be observed with chronic neonatal lung diseases associated with prematurity, chronic vascular remodeling due to pulmonary overcirculation from left-to-right shunting, or genetic predisposition [1••].
aPHT is traditionally considered a disease of term and late preterm infants, but recently, prematurity has also been identified as an independent risk factor [17, 18]. aPHT is recognized in 2–8% of preterm infants presenting with early RDS and up to 67% of preterm infants with severe RDS [17,18,19]. aPHT has also been recognized as a risk factor for cPHT [20]. The high incidence of parenchymal lung disease, sepsis exposure, physiological immaturity of the NO pathway, and immature gas exchange mechanisms all play key roles in the pathogenesis of aPHT in the preterm infant. Preterm infants with fetal growth restriction, exposure to prolonged rupture of membranes with varying degrees of pulmonary hypoplasia, and chorioamnionitis are also at a higher risk of developing aPHT. The abnormal physiologic consequences of PHT also contribute to delayed cardiopulmonary transition in premature infants [16••].
The entity of cPHT is characterized by a later presentation (> 4–6 weeks chronological age) with a gradual increase in PVR. Established cPHT is commonly associated with CLD [21]. Although the reported incidence of cPHT is 14–44%, with an association of 20–50% mortality [22, 23••, 24, 25] in the infants with recognized CLD [20, 26], recent evidence indicates that up to 20% of extremely low gestational age neonates without CLD will develop some degree of pHTN evidence by echocardiography during the neonatal period [20, 27]. Reduced vascular branching, altered pattern of vascular distribution, disruption of vascular signaling pathways, endothelial injury, and abnormal smooth muscle proliferation are some of the underlying mechanisms of the late increase in PVR [28]. Exacerbating factors include air-trapping induced abnormal stretch of small pulmonary arteries, atelectasis leading to constraining pulmonary vessels, and acute episodes of hypoxia and hypercarbia, all of which can induce pulmonary artery vasoconstriction and altered vessel morphology in premature neonates [21].
Cardiopulmonary consequences: the hemodynamic profile of PHT
PHT leads to an array of complex hemodynamic consequences [1••]. The clinical presentation depends on the onset and severity of the disease process. Awareness of the spectrum of cardiopulmonary consequences of aPHT and cPHT facilitates appropriate monitoring and guides anticipatory management [21]. With aPHT, there are different etiologies that affect the cellular mechanisms, but the hemodynamic profile often remains the same. The elevated mPAP that is present with sustained elevation of PVR-related aPHT leads to failure of gas exchange across the highly constricted pulmonary blood vessels; this manifests as cyanosis and oxygenation failure from significant ventilation-perfusion mismatch early in life. The underlying parenchymal lung disease may also dictate the extent of oxygenation difficulty. Profound lability is another characteristic finding that indicates unstable pulmonary vasoreactivity with worsening oxygenation that parallels increases in PVR with handling and agitation [29].
Neonatal PHT with persistent high PVR results in a direct increase in RV afterload [1••]. The neonatal myocardium is exquisitely sensitive to afterload and often incapable of rapid adaptation (this is even more pronounced in the preterm infant), perpetuating progressive RV dilatation and dysfunction [29]. The coexistence of high PVR and RV dysfunction can result in critically low PBF. The increased RV systolic pressure with high PVR is transmitted to the atrial level, leading to the reversal of shunt across the patent foramen ovale and intracardiac mixing of oxygenated and deoxygenated blood that clinically manifests as cyanosis. The high mPAP also drives a bidirectional or possibly complete right-to-left shunt across the DA, resulting in post-ductal mixing of deoxygenated blood and the classical presentation of differential cyanosis. Although a right-to-left shunt across the DA may offload the pulmonary circulation and mitigate some of the RV failure and systemic hypoperfusion, it can reduce the myocardial oxygen demand and lead to further ventricular dysfunction. LV filling may also be affected by reduced PBF, RV dilatation, and septal bowing. Varying degree of LV dysfunction may manifest due to the phenomenon of interventricular dependence in the context of RV dysfunction. The diversion of PBF away from the lungs may also cause acidosis and impede global myocardial performance, eventually leading to further LV dysfunction with subsequent decline in LV output and clinical manifestations of hypotension, shock, oliguria/anuria, and end-organ compromise [1••].
In contrast to the acute hypoxic respiratory failure and hemodynamic instability that characterizes classic aPHT, the symptoms and clinical findings from chronically elevated PAP are more subtle and insidious in cPHT [23••, 30]. The cPHT often manifests with oxygen dependence, rather than oxygenation failure, and respiratory deterioration in the face of expected improvement. Prolonged exposure to increased afterload in cPHT often leads to RV dysfunction, both systolic and diastolic, manifesting through a constellation of marked signs of progressive right heart failure including RV dilatation, dysfunction, hepatomegaly, edema, excessive weight gain, and/or inability to establish oral feeding [30].
Clinical and echocardiographic assessment of neonatal pulmonary hypertension
Hemodynamic appraisal of neonates with PHT begins with a comprehensive clinical history to identify pertinent maternal, perinatal, and postnatal influences that can increase the risk of cardiovascular compromise. Since objective measurement of cardiac output, systemic blood flow, and end-organ perfusion remain very challenging, assessment must combine various clinical parameters (i.e., oxygen saturations, continuous blood pressure, heart rate, urine output, capillary refill time, etc.), biochemical markers (lactate, base excess), and imaging modalities (e.g., echocardiography) to provide the most complete and accurate picture of the cause of hemodynamic instability, inform decision-making, and offer a possible therapeutic approach [29]. The use of echocardiography to assess for PHT and cardiovascular health in neonates has now become the standard of care [31, 32] with a growing recognition that it can provide hemodynamic information that either complements what is clinically suspected or delivers novel physiologic insight [29].
Cardiac catheterization remains the gold standard for the assessment of RV performance and pulmonary hemodynamics, but it remains invasive and less than ideal in neonates when searching for a modality to screen and monitor PHT in this population [33]. The integration of hemodynamic information obtained by transthoracic echocardiography relevant to the etiology and clinical situation offers a vehicle for which to formulate a scientifically based diagnostic impression, determine a pathophysiological choice for support, and evaluate the response to therapeutic intervention [29]. In order to provide a relevant blueprint for the diagnosis of both aPHT and cPHT, we suggest classifying commonly used measurements into three broad categories: (1) indirect assessment of elevated RV afterload, (2) estimation of pulmonary hemodynamics, and (3) measures of right ventricular performance. (Left ventricular performance is also important in the evaluation of neonatal PHT but beyond the scope of this review) (Table 1).
Indirect assessment of elevated RV afterload
There are several cardiopulmonary interactions that provide an indirect assessment of PHT shown by evidence of elevated RV afterload. The size and shape of the interventricular septum (IVS) can be a useful tool for qualitative assessment of elevated mPAP with the degree of flattening in end-systole providing an estimate of RV systolic pressure (RVSP) between 50 and 100% of the systemic pressures [34]. The eccentricity index (EI) is a measure that quantifies the ratio between LV anteroposterior and septolateral dimensions and suggests elevated RV afterload when this ratio is > 1.0 [35]. While LV diastolic EI is more of a marker of RV volume overload, systolic EI reflects RV pressure overload and is more useful for PHT [36•]. Evidence of elevated PAP can be associated with RV dilation in aPHT and RV hypertrophy in cPHT. Measuring systolic time intervals can provide insight to pulmonary vascular impedance, compliance, and pressure [33]. Pre-ejection period (PEP) is defined as the onset of ventricular depolarization to beginning of pulmonary ejection and is directly proportional to PAP [37]. RV ejection time (RVET) is relative to stroke volume and has an inverse relationship to pulmonary artery compliance. Pulmonary artery acceleration time (PAAT) is the time interval from the onset to the peak of ejection and provides a strong correlation to systolic PAP [33]. Ratios of these respective intervals such as PEP/RVET and PAAT/RVET have been shown to provide reliable estimates of invasive measures of pulmonary hemodynamics and RV performance in both acute and chronic PHT [33, 38].
Estimation of pulmonary hemodynamics
Doppler interrogation of the tricuspid and pulmonary flow is useful in estimating ventricular systolic pressures. In the setting of absent RV outflow tract obstruction, it can also predict pulmonary artery systolic pressure (PASP). In neonates with tricuspid valve regurgitation velocity (TRV), the modified Bernoulli equation can estimate RVSP by the following equations: 4 × (TRV)2 + right atrial pressure [39]. There are several limitations with this equation, but the trend is often valuable for following neonates with aPHT and cPHT. Pulmonary regurgitation velocity (PRV) has been shown to accurately measure both mPAP and end-diastolic pulmonary pressure by peak regurgitant velocity and end-diastolic regurgitant velocity respectively [40]. This is particularly useful to estimate the mean transpulmonary gradient in the calculation of PVR and distinguishing left-sided obstructive disease. Using TRV to estimate RVSP, pulmonary artery diastolic pressure (PADP) can also be estimated by the following equation: PADP = 0.49 × RVSP [41]. Velocities across a VSD and a PDA can also be used to estimate pulmonary pressures depending on the location, length, and tortuosity.
PVR can be assessed with the TRV interrogation based on its relationship to velocity time integral along the RV outflow tract (TRV:VTI) [42], dynamic compliance [43], and pulmonary artery capacitance [44], all of which add value to understanding the degree of pulmonary bed vasoreactivity. PVR can also be assessed by measuring RV systolic time intervals (RVET, PAAT, PEP), as they have been shown to provide accurate estimates of invasive measures of PVR and pulmonary artery compliance [33, 38].
Right ventricular performance
The complex cardiopulmonary interactions and myocardial remodeling seen in both aPHT and cPHT result in varying degrees of systolic and diastolic ventricular dysfunction. RV output (RVO) provides an estimate of PBF in the absence of RV outflow obstructions or pulmonary-to-aortic shunts. Although there are limitations and considerations with proper acquisition, serial measurements may provide insight into alterations in stroke volume and PBF-related PHT. The geometric shape, fiber orientation, and coarse trabeculations make estimating contractile function and defining ventricular borders difficult with conventional 2D echocardiography. Several novel and re-emerging measures have been validated to overcome these concerns. Percent fractional area change (FAC) is a two-dimensional measurement of surface area used to characterize RV global systolic function and has been well studied in preterm and term infants [45, 46]. Tricuspid annular plane systolic excursion (TAPSE) measures the longitudinal excursion of the tricuspid valve from the base towards the apex during systole [47]. Although the RV has a complex three-dimensional myofiber arrangement, the dominant longitudinal shortening measured by TAPSE provides the major contribution to ejection fraction and stroke volume during systole [46]. In addition, both reduced values of FAC and TAPSE have been found to be predictive of increased mortality in patients with PHT [48]. The relationship of TAPSE to PAAT has recently been shown to be inversely correlated with mPAP and PVR and directly correlated with pulmonary arterial compliance and RV strain in a cohort of children [49]. TAPSE:PAAT provides a reliable estimate of RV-pulmonary vascular coupling [50].
Other measures of RV performance, such as the systolic and diastolic time (SD/DD) and RV myocardial performance index (MPI) are markers of global systolic and diastolic function reflecting ventricular loading and contractility. An increase in SD/DD ratio is seen as a sign of global RV dysfunction secondary to increased afterload; a level of > 1.3 has been associated with the need for extracorporeal membrane oxygenation (ECMO) or death [51]. RV MPI relates the sum of isovolumic contraction and relaxation time to the ejection time with prolonged isovolumic intervals; higher MPI values are also suggestive of increased RV afterload and ventricular dysfunction [51]. Tissue Doppler imaging measures the peak systolic (s’), early diastolic (e’), late diastolic (a’), and peak isovolumetric contraction velocities [52]. Normative values exist in neonates and can provide longitudinal measurements of RV diastolic performance in preterm infants [8]. Speckle-tracking echocardiography is used to assess global and free wall longitudinal strain and has been shown to be a predictive measure of PHT in neonates and children [53].
Morphological measures of RV performance can also provide diagnostic clues for RV hypertrophy and dilation. Structural assessments by echocardiography should include measures of areas (end systolic and end diastolic) [45], cavity dimensions at the base, mid-cavity, and length of the RV from the apex to the middle of the base in the RV-focused apical four-chamber view. RV outflow dimensions can be obtained from either the parasternal long axis or short axis view to assess the proximal and distal components of the RV outflow tract [54].
Management of pulmonary hypertension
Management of aPHT and cPHT both require attention to physiology with the aim of promoting adequate PBF to improve the efficacy of oxygenation and reduce secondary consequences of increased RV afterload [1••]. The initial management for PHT begins with the identification of risk factors, recognition of symptoms, and anticipation of potential illness. The approach consists of three basic tenants: (1) supportive cardiorespiratory care, (2) judicious use of pulmonary vasodilators, and/or (3) invasive extracorporeal membrane oxygenation if needed.
General cardiorespiratory measures may reverse or prevent further increase in pulmonary vasoconstriction and include the following: (i) correction of metabolic derangements, including hypoglycemia, hypothermia, and acidosis; (ii) maintenance of adequate oxygenation with supplemental oxygen as the initial therapy in both aPHT and cPHT. Although the target oxygen concentration ideal for optimizing outcomes for neonates is not established, recent recommendations consider the goal to maintain oxygen saturations between 92 and 95% [23••, 55]; (iii) optimization of lung recruitment with mechanical ventilation guided by the underlying mechanism of PHT and response to treatment. In cPHT, ventilator strategies should have a specific emphasis on avoiding air trapping and minimizing acidosis. In the setting of hypoxemia caused by right-to-left shunting, rather than ventilation-perfusion imbalance, minimizing mean airway pressure while maintaining adequate lung recruitment and ensuring venous return may be most appropriate. Surfactant replacement therapy can be considered for infants with aPHT and pulmonary diffusion impairment [56], but early refractory hypoxemia despite effective ventilation strategies and targeted surfactant administration may relate to extreme lung immaturity in premature infants, with PH as a contributing factor. (iv) Circulatory support with fluid resuscitation, inotropic agents, or vasopressors to ensure adequate cardiac output and systemic perfusion may be necessary with aPHT. While specific medications and their mechanisms of action are beyond the scope of this review, steroid treatment to stabilize blood pressure in inotropic-resistant environments may also be necessary. In addition to circulatory support, steroids may also mitigate the chemical pneumonitis seen with meconium aspiration in aPHT with the inhibition of inflammation and decreasing cytokine-induced vasoconstriction. Laboratory evidence and clinical observations also suggest that modulating inflammation using glucocorticoids may benefit neonates with cPHT [57]; (v) sedating agents should be considered in an effort to avoid agitation and the catecholamine release that contributes to further increases in afterload and hypoxemia from right-to-left shunting and asynchronous ventilation with patient-triggered ventilator mode.
Additional cardiorespiratory support measures may also be considered based on the physiologic presentation. They include the administration of packed red blood cell transfusion for the optimization of oxygen delivery [58] and antibiotic treatment with acute episodes of neonatal PHT that can occur with sepsis physiology. Since pulmonary edema may also be a major contributor to the symptoms of cPHT, diuretics may be considered when cardiac preload is adequate and are especially important in the setting of shunt lesions [23••, 30]. In flow-related PHT, flow modulation strategies (e.g., medical/surgical closure of DA or intracardiac shunt) may be required. In addition to appropriate ventilation strategies, assessments for aspiration and structural airway disease should be performed prior to considering PHT-targeted therapy.
In severe cases, general supportive care may be insufficient to maintain adequate oxygenation. PHT-targeted therapy should be considered for infants with sustained PHT after optimal treatment of underlying respiratory and cardiac disease [23••]. Inhaled NO (iNO), a potent and microselective pulmonary vasodilator, and ECMO are two therapeutic options with scientifically proven benefits for infants with PHT who fail to respond to general cardiopulmonary supportive care [59]. In addition, several newer non-invasive therapeutic agents have been developed, many of which are used routinely in clinical practice for children and adults with PHT, but their efficacy and safety have not been tested in large clinical trials in neonates [23••]. One way to approach the different therapeutic options for PHT is to think of them with respects to the key mediator pathways listed above. Oxygen is the starting point for all pathways [1••]. The NO-cGMP pathway has two recognized therapeutic agents, iNO and sildenafil, for the treatment of PHT in neonates. iNO is indicated in mechanically ventilated term and late preterm newborns if PaO2 < 100 mmHg or OI > 25 [55, 56]. Although it has been studied and proven ineffective for the prevention of CLD in premature infants, a trial of iNO may be considered in those at risk for PHT and/or echocardiographic evidence of PHT beyond what is expected. Response to iNO in premature infants has been shown to be improved in the presence of echocardiographic evidence of aPHT [19]. Sildenafil, a phosphodiesterase type 5 inhibitor, reduces PVR; it may be considered in this population when iNO is not available, PHT is refractory to iNO (e.g., OI > 25) or with weaning from iNO [23••, 55]. The prostacyclin-cAMP pathway also has two primary therapeutic options: prostacyclin (PGI2) agonists and milrinone. PGI2 can be administered via an intravenous, inhaled, or subcutaneous route and can provide both systemic and pulmonary vasodilatation effects depending on the route and dose. Milrinone, a selective phosphodiesterase 3 (PDE3) inhibitor, causes the relaxation of vascular smooth muscle and, in some settings, can enhance myocardial contractility (inotropy) and improve myocardial relaxation (lusitropy). The endothelin toxin 1 pathway can be mediated by PO administration of bosentan, a non-selective endothelin receptor antagonist.
If echocardiography evaluation demonstrates RV dysfunction in infants with hemodynamic compromise and PHT physiology, treatment strategies should be tailored to the pathogenic contributions to altered RV performance; (1) decreased preload with volume optimization, (2) increased PVR with RV afterload reduction, and/or (3) altered contractility with RV inotropy enhancement [1••]. Prostaglandins (PgE1) to re-open or maintain the DA in aPHT may also be considered in those neonates with PHT physiology but without a pathway to offload the RV. The fulminant presentation of aPHT often necessitates a rapid and multipronged approach. The cornerstone of its management is optimal lung parenchymal support followed by early pulmonary vasodilator therapy, which in most cases will show satisfactory response. On the other hand, because of the insidious nature and underlying pathophysiology of cPHT, the primary treatment focus is on mitigating ongoing lung injury, optimizing respiratory support, promoting growth and nutrition, and supporting RV function (e.g., with preload reduction) rather than use of pulmonary vasodilators.
Abbreviations
- aPHT:
-
Acute pulmonary hypertension
- CAMP:
-
Cyclic adenosine monophosphate
- CDH:
-
Congenital diaphragmatic hernia
- CGMP:
-
Cyclic guanylyl monophosphate
- CLD:
-
Chronic lung disease
- cPHT:
-
Chronic pulmonary hypertension
- DA:
-
Ductus arteriosus
- EI:
-
Eccentricity index
- FAC:
-
Fractional area change
- HIE:
-
Hypoxic ischemic events
- IDM:
-
Infants of diabetic mothers
- iNO:
-
Inhaled nitric oxide
- LV:
-
Left ventricle
- mPAP:
-
Mean pulmonary artery pressure
- MPI:
-
Myocardial performance index
- PAAT:
-
Pulmonary artery acceleration time
- PAP:
-
Pulmonary artery pressure
- PASP:
-
Pulmonary artery systolic pressure
- PADP:
-
Pulmonary artery diastolic pressure
- PCWP:
-
Pulmonary capillary wedge pressure
- PBF:
-
Pulmonary blood flow
- PDA:
-
Patent ductus arteriosus
- PEP:
-
Pre-ejection period
- PGI2 :
-
Prostacyclin
- PHT:
-
Pulmonary hypertension
- PRV:
-
Pulmonary regurgitation velocity
- RDS:
-
Respiratory distress syndrome
- RV:
-
Right ventricle
- RVET:
-
Right ventricle ejection time
- RVO:
-
Right ventricular output
- SVR:
-
Systemic vascular resistance
- TR:
-
Tricuspid regurgitation
- TRV:
-
Tricuspid valve regurgitation velocity
- TAPSE:
-
Tricuspid annular plane systolic excursion
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
•• Jain A, McNamara PJ. Persistent pulmonary hypertension of the newborn: advances in diagnosis and treatment. Semin Fetal Neonatal Med. 2015;20:262–71 This article provides a comprehensive overview of the pathological contributors to PHT, the physiologic constituents of its phenotypic expression, standard approach to therapeutic intervention, and the role of bedside echocardiography in enhancing the decision-making process.
Haddad F, Hunt SA, Rosenthal DN, Murphy DJ. Right Ventricular function in cardiovascular disease, part I: anatomy, physiology, aging, and functional assessment of the right ventricle. Circulation. 2008;117:1436–48.
Paige SL, Plonowska K, Xu A, Wu SM. Molecular regulation of cardiomyocyte differentiation. Circ Res. 2015;116:341–53.
Mielke G, Benda N. Cardiac output and central distribution of blood flow in the human fetus. Circulation. 2001;103:1662–8.
Hendson L, Emery DJ, Phillipos EZ, Bhargava R, Olley PM, Lemke RP. Persistent pulmonary hypertension of the newborn presenting as the primary manifestation of intracranial arteriovenous malformation of the vein of Galen. Am J Perinatol. 2000;17:405–10.
Tiwary S, Geethanath RM, Abu-Harb M. Vein of Galen malformation presenting as persistent pulmonary hypertension of newborn (PPHN). BMJ Case Rep. 2013.
Rothstein R, Paris Y, Quizon A. Pulmonary hypertension. Pediatr Rev. 2009;30:39–45.
Bussmann N, El-Khuffash A, Breatnach CR, McCallion N, Franklin O, Singh GK, et al. Left ventricular diastolic function influences right ventricular — pulmonary vascular coupling in premature infants. Early Hum Dev. 2018;128:35–40.
Giesinger RE, Bailey LJ, Deshpande P, McNamara PJ. Hypoxic-ischemic encephalopathy and therapeutic hypothermia: the hemodynamic perspective. J Pediatr. 2017;180:22–30.
Drossner DM, Kim DW, Maher KO, Mahle WT. Pulmonary vein stenosis: prematurity and associated conditions. Pediatrics. 2008;122:e656–61.
Laudy JAM, Wladimiroff JW. The fetal lung 1: developmental aspects. Ultrasound Obstet Gynecol. 2000;16:284–90.
Prsa M, Sun L, van Amerom J, Yoo SJ, Grosse-Wortmann L, Jaeggi E, et al. Reference ranges of blood flow in the major vessels of the normal human fetal circulation at term by phase-contrast magnetic resonance imaging. Circ Cardiovasc Imaging. 2014;7:663–70.
Lakshminrusimha S, Steinhorn RH. Pulmonary vascular biology during neonatal transition. Clin Perinatol. 1999;26:601–19.
Puthiyachirakkal M, Mhanna MJ. Pathophysiology, management, and outcome of persistent pulmonary hypertension of the newborn: a clinical review. Front Pediatr. 2013;1:23.
Abman SH, Chatfield BA, Rodman DM, Hall SL, McMurtry IF. Maturational changes in endothelium-derived relaxing factor activity of ovine pulmonary arteries in vitro. Am J Phys. 1991;260(4 Pt 1):L280–5.
•• Giesinger RE, More K, Odame J, Jain A, Jankov RP, McNamara PJ. Controversies in the identification and management of acute pulmonary hypertension in preterm neonates. Pediatr Res. 2017;82:901–14 Provides a diagnostic approach for acute hypoxemic respiratory failure in the preterm neonate, outlines the pathophysiological conditions that may present as aPHT, and discusses the implications of high pulmonary vascular resistance (PVR) on the cardiovascular system.
Nakanishi H, Suenaga H, Uchiyama A, Kusuda S. Persistent pulmonary hypertension of the newborn in extremely preterm infants: a Japanese cohort study. Arch Dis Child Fetal Neonatal Ed. 2018;103(6):F554–f61.
Steurer MA, Jelliffe-Pawlowski LL, Baer RJ, Partridge JC, Rogers EE, Keller RL. Persistent pulmonary hypertension of the newborn in late preterm and term infants in California. Pediatrics. 2017;139.
Dani C, Corsini I, Cangemi J, Vangi V, Pratesi S. Nitric oxide for the treatment of preterm infants with severe RDS and pulmonary hypertension. Pediatr Pulmonol. 2017;52:1461–8.
Mourani PM, Sontag MK, Younoszai A, Miller JI, Kinsella JP, Baker CD, et al. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2015;191:87–95.
Mourani PM, Abman SH. Pulmonary hypertension and vascular abnormalities in bronchopulmonary dysplasia. Clin Perinatol. 2015;42:839–55.
Berenz A, Vergales JE, Swanson JR, Sinkin RA. Evidence of early pulmonary hypertension is associated with increased mortality in very low birth weight infants. Am J Perinatol. 2017.
•• Krishnan U, Feinstein JA, Adatia I, Austin ED, Mullen MP, Hopper RK, et al. Evaluation and management of pulmonary hypertension in children with bronchopulmonary dysplasia. J Pediatr. 2017;188:24–34 Provides detailed consensus recommendations for the care of children with BPD-PHT.
Murthy K, Dykes FD, Padula MA, Pallotto EK, Reber KM, Durand DJ, et al. The Children’s Hospitals Neonatal Database: an overview of patient complexity, outcomes and variation in care. J Perinatol. 2014;34:582–6.
Khemani E, McElhinney DB, Rhein L, Andrade O, Lacro RV. Thomas KC Pulmonary artery hypertension in formerly premature infants with bronchopulmonary dysplasia: clinical features and outcomes in the surfactant era. Pediatrics. 2007;120:1260–9.
Kwon HW, Kim HS, An HS, Kwon BS, Kim GB, Shin SH, et al. Long-term outcomes of pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Neonatology. 2016;110:181–9.
Weismann CG, Asnes JD, Bazzy-Asaad A, Tolomeo C, Ehrenkranz RA, Bizzarro MJ. Pulmonary hypertension in preterm infants: results of a prospective screening program. J Perinatol. 2017;37:572–7.
De Paepe ME, Mao Q, Powell J, Rubin SE, DeKoninck P, Appel N, et al. Growth of pulmonary microvasculature in ventilated preterm infants. Am J Respir Crit Care Med. 2006;173:204–11.
El-Khuffash A, McNamara PJ. Hemodynamic assessment and monitoring of premature infants. Clin Perinatol. 2017;44(2):377–93.
Neary E, Jain A. Right ventricular congestion in preterm neonates with chronic pulmonary hypertension. J Perinatol. United States 2018.
El-Khuffash AF, McNamara PJ. Neonatologist-performed functional echocardiography in the neonatal intensive care unit. Semin Fetal Neonatal Med. 2011;16:50–60.
Evans N, Gournay V, Cabanas F, Kluckow M, Leone T. Groves A Point-of-care ultrasound in the neonatal intensive care unit: international perspectives. Semin Fetal Neonatal Med. 2011;16:61–8.
Levy PT, Patel MD, Groh G, Choudhry S, Murphy J, Holland MR, et al. Pulmonary artery acceleration time provides a reliable estimate of invasive pulmonary hemodynamics in children. J Am Soc Echocardiogr. 2016;29:1056–65.
Kim GB. Pulmonary hypertension in infants with bronchopulmonary dysplasia. Korean J Pediatr. 2010;53:688–93.
Ehrmann DE, Mourani PM, Abman SH, Poindexter BB, Morrow LA, Wagner BD, et al. Echocardiographic measurements of right ventricular mechanics in infants with bronchopulmonary dysplasia at 36 weeks postmenstrual age. J Pediatr. 2018;203:10–21.
• de Boode WP, Singh Y, Molnar Z, Schubert U, Savoia M, Sehgal A. Application of neonatologist performed echocardiography in the assessment and management of persistent pulmonary hypertension of the newborn. Pediatr Res. 2018;84(Suppl 1):68–77 Explore the applications of neonatal performed echocardiography techniques that aid in the correct diagnostic and pathophysiological assessment of the most common neonatal etiologies of PHT and provide guidelines for using these techniques to optimize the management.
Hsieh KS, Sanders SP, Colan SD, MacPherson D, Holland C. Right ventricular systolic time intervals: comparison of echocardiographic and Doppler-derived values. Am Heart J. 1986;112:103–7.
Jain A, Mohamed A, Kavanagh B, Shah PS, Kuipers BCW, El-Khuffash A. Cardiopulmonary adaptation during first day of life in human neonates. J Pediatr. 2018;200:50–57.e2.
Yock PG, Popp RL. Noninvasive estimation of right ventricular systolic pressure by Doppler ultrasound in patients with tricuspid regurgitation. Circulation. 1984;70:657–62.
Abbas AE, Fortuin FD, Schiller NB, Appleton CP, Moreno CA, Lester SJ. Echocardiographic determination of mean pulmonary artery pressure. Am J Cardiol. 2003;92:1373–6.
Friedberg MK, Feinstein JA, Rosenthal DN. A novel echocardiographic Doppler method for estimation of pulmonary arterial pressures. J Am Soc Echocardiogr. 2006;19:559–62.
Abbas AE, Fortuin FD, Schiller NB, Appleton CP, Moreno CA, Lester SJ. A simple method for noninvasive estimation of pulmonary vascular resistance. J Am Coll Cardiol. 2003;41:1021–7.
Dyer K, Lanning C, Das B, Lee PF, Ivy DD, Valdes-Cruz L. Noninvasive Doppler tissue measurement of pulmonary artery compliance in children with pulmonary hypertension. J Am Soc Echocardiogr. 2006;19:403–12.
Mahapatra S, Nishimura RA, Oh JK, McGoon MD. Noninvasive assessment of pulmonary arterial capacitance by echocardiography. J Am Soc Echocardiogr. 2006;19:1045–50.
Levy PT, Dioneda B, Holland MR, Sekarski TJ, Lee CK, Mathur A, et al. Right ventricular function in preterm and term neonates: reference values for right ventricle areas and fractional area of change. J Am Soc Echocardiogr. 2015;28:559–69.
Breatnach CR, Levy PT, James AT, Franklin O, El-Khuffash A. Novel echocardiography methods in the functional assessment of the newborn heart. Neonatology. 2016;110:248–60.
Lammers AE, Haworth SG, Riley G, Maslin K, Diller GP, Marek J. Value of tissue Doppler echocardiography in children with pulmonary hypertension. J Am Soc Echocardiogr. 2012;25:504–10.
Malowitz JR, Forsha DE, Smith PB, Cotten CM, Barker PC, Tatum GH. Right ventricular echocardiographic indices predict poor outcomes in infants with persistent pulmonary hypertension of the newborn. Eur Heart J Cardiovasc Imaging. 2015;16:1224–31.
Levy PT, El Khufash A, Woo KV, Hauck A, Hamvas A, Singh GK. Right ventricular-pulmonary vascular interactions: an emerging role for pulmonary artery acceleration time by echocardiography in adults and children. J Am Soc Echocardiogr. 2018;31:962–4.
Levy PT, El Khufash A, Woo KV, Singh GK. A Novel noninvasive index to characterize right ventricle pulmonary arterial vascular coupling in children. JACC Cardiovasc Imaging. 2018
Aggarwal S, Natarajan G. Echocardiographic correlates of persistent pulmonary hypertension of the newborn. Early Hum Dev. 2015;91:285–9.
Jain A, Mohamed A, El-Khuffash A, Connelly KA, Dallaire F, Jankov RP, et al. A comprehensive echocardiographic protocol for assessing neonatal right ventricular dimensions and function in the transitional period: normative data and z scores. J Am Soc Echocardiogr. 2014;27:1293–304.
Levy PT, El-Khuffash A, Patel MD, Breatnach CR, James AT, Sanchez AA, et al. Maturational patterns of systolic ventricular deformation mechanics by two-dimensional speckle tracking echocardiography in preterm infants over the first year of age. J Am Soc Echocardiogr. 2017;30:685–98.
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28:1–39.
Abman SH, Hansmann G, Archer SL, Ivy DD, Adatia I, Chung WK, et al. Pediatric pulmonary hypertension: guidelines from the American Heart Association and American Thoracic Society. Circulation. 2015;132:2037–99.
Hilgendorff A, Apitz C, Bonnet D, Hansmann G. Pulmonary hypertension associated with acute or chronic lung diseases in the preterm and term neonate and infant. The European Pediatric Pulmonary Vascular Disease Network, endorsed by ISHLT and DGPK. Heart. 2016;102(Suppl 2):ii49–56.
Aggarwal M, Grady RM. Glucocorticoids for treating pediatric pulmonary hypertension: a novel use for a common medication. Cardiol Young. 2017;27:1410–2.
Mathew R, Huang J, Wu JM, Fallon JT, Gewitz MH. Hematological disorders and pulmonary hypertension. World J Cardiol. 2016;8:703–18.
Finer NN, Barrington KJ. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev. 2006;4:CD000399.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Pediatric and Congenital Heart Disease
Rights and permissions
About this article

Cite this article
Bhattacharya, S., Sen, S., Levy, P.T. et al. Comprehensive Evaluation of Right Heart Performance and Pulmonary Hemodynamics in Neonatal Pulmonary Hypertension. Curr Treat Options Cardio Med 21, 10 (2019). https://doi.org/10.1007/s11936-019-0713-8
Published:
DOI: https://doi.org/10.1007/s11936-019-0713-8