
Abstract
Lipoprotein(a) (Lp(a)) is an internationally accepted independent atherogenic risk factor. Details about its synthesis, many aspects of composition and clearance from the bloodstream are still unknown. LDL receptor (LDLR) (and probably other receptors) play a role in the elimination of Lp(a) particles. Proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors increase the number of available LDLRs and in this way very effectively reduce the LDL cholesterol (LDL-C) concentrations. As shown in controlled studies using PCSK9 inhibitors, Lp(a) levels are decreased by 20 to 30%, though in some patients no effect was observed. So far, it has not been clarified whether this decrease is associated with an effect on the incidence of cardiovascular events (CVEs). In two recently published well-performed secondary prevention studies (FOURIER with evolocumab, ODYSSEY OUTCOMES with alirocumab) baseline Lp(a) levels were shown to have an impact on CVEs independently of baseline LDL-C concentrations. The rather modest PCSK9 inhibitor-induced decrease of Lp(a) was associated with a reduction of CVEs in both studies, even after adjusting (ODYSSEY OUTCOMES) for demographic variables (age, sex, race, region), baseline Lp(a), baseline LDL-C, change in LDL-C, and clinical variables (time from acute coronary syndrome, body mass index, diabetes, smoking history). The largest decrease of CVEs was seen in patients with relatively low concentrations of both LDL-C and Lp(a) (FOURIER). These findings will probably have an influence on the use of PCSK9 inhibitors in patients with high Lp(a) concentrations.
Similar content being viewed by others

Lipoprotein(a)—synthesis, composition, metabolism, and clinical significance
Lipoprotein(a) (Lp(a)) consists of an LDL particle to which an apolipoprotein(a) (apo(a)) is linked with a single disulfide bond. The binding between apolipoprotein (B) (apoB), the major apolipoprotein of the LDL, and apo(a) takes place either in the hepatic cells, in the space of Disse, or in the vascular lumen [1]. The cholesterol content of the LDL in Lp(a) varies between 30 and 45%. The Lp(a) concentration is genetically determined. Mutations in the Lp(a) gene (LPA) and especially a variable number of LPA kringles IV type 2 in the apo(a) have an effect. A low number of these kringles is associated with higher Lp(a) levels.
The clearance of Lp(a) from the bloodstream is still not fully understood. Hepatic (LDL receptor (LDLR), VLDL receptor, scavenger receptor B1, LDL receptor–related protein 1, cluster of differentiation 36 receptor (CD36), plasminogen receptor) and nonhepatic receptors are probably involved [1,2,3,4]. Renal mechanisms may also play a role.
It is assumed that LDLRs only play a significant role in Lp(a) clearance when hepatic levels of the receptor are very high and LDL-C levels are low, as is the case in proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitor therapy. It is possible that apo(a) isoform length influences clearance behavior of Lp(a) in human plasma following upregulation of LDLRs.
The physiological significance of Lp(a) particles may relate to their procoagulatory properties. Thus, wound healing could be stimulated.
On the other hand, Lp(a) induces atherosclerotic lesions and is supposed to promote aortic valve stenosis: a combination of proatherosclerotic, proinflammatory and procoagulatory actions seems to be responsible [5]. Data suggest that the atherogenicity of Lp(a) may be mediated in part by proinflammatory oxidized phospholipids [6].
Plasma PCSK9 is found in association with Lp(a) particles in humans with high Lp(a) levels and in mice carrying human Lp(a) [7].
Studies using epidemiological data, Mendelian randomization and genome-wide associations have proven that elevated Lp(a) induce cardiovascular events (CVEs) like myocardial infarction (MI), stroke, occlusions of carotids or of leg arteries [5, 8].
New data obtained in the Danish population with respect to the relationship between Lp(a) and mortality have recently been published: In this study Lp(a) levels >93 mg/dl (199 nmol/l; 96th–100th percentiles) versus <10 mg/dl (18 nmol/l; 1st–50th percentiles) were associated with a hazard ratio (HR) of 1.50 (95% CI 1.28–1.76) for cardiovascular mortality and of 1.20 (1.10–1.30) for all-cause mortality [9]. High levels of Lp(a), induced by low LPA kringle IV type 2 number of repeats rather than through high cholesterol content, were associated with increased mortality.
Lp(a) levels in the atherothrombotic range are generally accepted as >30 to 50 mg/dl or >75 to 125 nmol/l [1]. Such levels affect 20 to 30% of the global population, with possibly higher incidence in patients with established cardiovascular disease and calcific aortic valve disease.
PCSK9 inhibitors—mode of action
PCSK9 inhibitors are human monoclonal antibodies binding to the PCSK9 protein. This protein binds to LDLRs for endocytosis and lysosome degradation in the liver, resulting in an increase in circulating LDL cholesterol (LDL-C) level. LDLRs usually recycle after they transported LDL particles into the cells. By inhibiting this LDLR destruction, the number of LDLRs at the cell surface markedly increases—leading to an effective removal of circulating LDL particles. Thus, reductions of LDL-C of more than 50% can be reached.
Evidently, the PCSK9 protein also exerts effects on other receptors like VLDL receptor, LDL receptor-related protein 1 or the apolipoprotein E receptor [3].
It was shown that PCSK9 may increase the secretion of apo(a) and of apoB—an inhibition of PCSK9 may counteract these effects [10]. Thus PCSK9 inhibitors reduce Lp(a) levels by both increasing clearance and reducing its synthesis.
Lp(a) kinetics were studied using intravenous D3-leucine administration, mass spectrometry, and compartmental modeling [11]. Evolocumab monotherapy was shown to lower the plasma Lp(a) pool size by decreasing the production of Lp(a) particles. In combination with atorvastatin, evolocumab lowered the plasma Lp(a) pool size by accelerating the catabolism of Lp(a) particles.
Effects of PCSK9 inhibitors on Lp(a) concentrations
Meta-analysis of randomized controlled trials with PCSK9 inhibitors
A meta-analysis of studies using PCSK9 inhibitors (27 randomized controlled trials [RCTs] in 11,864 patients) documented a mean reduction of Lp(a) levels by 21.90% (95% CI 24.28, 19.51) [10]. Treatment modalities like type of PCSK9 inhibitor, duration of therapy, application in patients with or without familial hypercholesterolemia, monotherapy or combination therapy, types of control treatment (placebo, ezetimibe), baseline Lp(a) (below 50 or above 50 mg/dl), and immunoassay did not have a significant influence on this reduction. The lower the reached LDL-C concentration was, the higher the reduction rate for Lp(a). Attention should be paid to the fact that in the majority of the included studies mean Lp(a) ranged between 9 and 40 mg/dl; however, in one investigation this value was 100 (but SD 162!) mg/dl.
Evolocumab in patients with very high Lp(a) concentrations
In 65 patients with Lp(a) concentrations of about 200 nmol/l an injection therapy with 420 mg evolocumab once in 4 weeks reduced Lp(a) after 16 weeks by 28.0 (56.5, 9.0) nmol/l (median, IQR), equivalent to 13.9 (19.3, 8.5) percent (mean, 95% CI) [12]. LDL-C was decreased by 2.2 (0.8) mmol/l, corresponding to 60.7 (65.8, 55.5) percent (mean, 95% CI).
Interestingly, arterial wall inflammation (most diseased segment target-to-background ratio (MDS TBR)) in the index vessel (left carotid, right carotid, or thoracic aorta) was not changed by evolocumab. The explanation of the authors for this lack of change is that Lp(a) levels remained high despite the rather modest reduction induced by the PCSK9 inhibitor.
Effect of PCSK9 inhibitors in patients undergoing lipoprotein apheresis therapy
ODYSSEY ESCAPE Study
Alirocumab (150 mg biweekly, 2:1 allocation to verum and placebo, respectively) was given to patients (n =62) who were treated with lipoprotein apheresis (LA) in order to evaluate the possibility to replace LA therapy by the PCSK9 inhibitor [13]. Alirocumab reduced Lp(a) levels in those patients with normal values (mean below 20 mg/dl) after 6 weeks by 15% (placebo controlled) and after 18 weeks by 2.7%. But in patients with high baseline Lp(a) concentrations (mean above 90 mg/dl) the corresponding changes amounted to 13% and to +1.9% (placebo controlled).
Observations of the authors
At our center we initiated PCSK9 inhibitor therapy in patients undergoing LA treatment when LDL-C levels remained high despite maximally tolerated lipid-lowering therapy (drugs, LA). Fig. 1 shows clear differences between patients with respect to lowering of LDL-C and Lp(a) (12 weeks after start of the injection therapy), indicating that reporting mean values is of limited significance when describing the effectiveness of PCSK9 inhibitors.

Individual percent reductions of LDL-C (a, n =41) and of Lp(a) (b, n =23) after 12 weeks of PCSK9 inhibitor application (usually biweekly) in patients on LA therapy. LDL‑C LDL cholesterol, Lp(a) Lipoprotein(a)
Among the 41 patients who started PCSK9 inhibitor therapy, only 23 showed elevated Lp(a) levels (higher than 120 nmol/l before first LA session).
Outcome data in prospective controlled PCSK9 inhibitor studies—association with the effects of these drugs on Lp(a) levels
FOURIER Study
The randomized FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Patients with Elevated Risk) Study tested the effect of evolocumab on cardiovascular outcomes versus placebo in patients with established atherosclerotic cardiovascular disease (median follow-up 2.2 years) [14].
Lp(a) was measured in 25,096 patients [15]. The median (IQR) baseline Lp(a) concentration was 37 (13, 165) nmol/l. In the placebo arm, patients with baseline Lp(a) in the highest quartile had a higher risk of coronary heart disease (CHD) death, MI or urgent revascularization (UR) (adjusted HR Q4:Q1 1.22, 95% CI 1.01,1.48) independent of LDL-C. At 48 weeks, evolocumab significantly reduced Lp(a) by a median (IQR) of 26.9% (6.2, 46.7%)—equivalent to 11 nmol/l (1, 32) absolute change. It is important to note that in more than 30% of the patients receiving evolocumab no reduction of Lp(a) was seen!
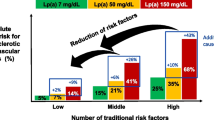
The percent change in Lp(a) and LDL-C at 48 weeks in evolocumab patients was moderately positively correlated. Evolocumab reduced the risk of CHD death, MI or UR by 23% (HR 0.77, 95% CI 0.67, 0.88) in patients with a baseline Lp(a) > median, and by 7% (HR 0.93, 0.80, 1.08) in those ≤ median. Coupled with the higher baseline risk, the absolute risk reductions and number-needed-to-treat for 3 years (NNT3y) were 2.49% and 40 vs. 0.95% and 105, respectively.
When a clinical threshold of 120 nmol/l (50 mg/dl) was applied, the absolute risk reductions and NNT3y were 2.41% and 41 for those above the threshold versus 1.41% and 71 below the threshold.
In a weighted least square linear regression analysis that examined the association between treatment effect on CHD death, MI or UR and per unit decrease in Lp(a) adjusting for differences in LDL-C, there was a significant relationship with a 15% relative risk reduction (95% CI 2, 26%, P = 0.0199) per 25 nmol/l reduction in Lp(a).
The authors observed a stepwise decrease in the risk of CHD death, MI or UR for patients who achieved either an Lp(a) or LDL-C value below the achieved median with the lowest event rate observed for those who achieved lower levels of both values. Compared with patients above the median achieved level for both lipid parameters, patients with at least one level below the median had a 15% lower risk of major coronary events (adjusted HR 0.85, 95% CI 0.75, 0.97, P = 0.01) and those with both levels below their respective medians had a 29% lower risk of major coronary events (adjusted HR 0.72, 95% CI 0.62, 0.83, P < 0.0001). It was reported that consistent results were observed when patients were stratified by achieved values of LDL-C 70 mg/dl and Lp(a) 120 nmol/l.
The incidence (3y KMrate, %) of the endpoint CHD death, MI and UR beyond week 12 was different depending on whether the reached Lp(a) and LDL-C levels where above or below the median (Table 1; [15]).
ODYSSEY OUTCOMES Trial
This is a multicenter, randomized, double-blind, placebo-controlled trial involving 18,924 patients who had an acute coronary syndrome 1 to 12 months earlier, had an LDL-C of at least 1.8 mmol/l (70 mg/dl), a non-high-density lipoprotein cholesterol level of at least 2.6 mmol/l (100 mg/dl), or an apoB level of at least 80 mg/dl, and were receiving statin therapy at a high-intensity dose or at the maximum tolerated dose [16]. Patients were randomly assigned to receive alirocumab subcutaneously or matching placebo every 2 weeks.
Data on Lp(a) have been presented at the ISA congress in Toronto (2018). The median level was 21.2 mg/dl (IQR 6.7, 59.6). Major CVEs occurred in the baseline 4th quartile more often than in the 1st quartile (HR unadjusted 1.37; adjusted (for age, sex, race, geographic region, time since event, BMI, smoking history, diabetes, baseline LDL-C) 1.28). A similar relationship was observed for non-fatal MI. No relationship was found with stroke, cardiovascular death, or all-cause death. In the course of the study alirocumab reduced Lp(a) levels in the mean by about 5 mg/dl. After 4 months these reductions amounted to 9.8 mg/dl (median; IQR 3.18, 16.2) in the 3rd quartile and to 20.2 mg/dl in the 4th quartile (median; IQR 8.0, 34.3), more than in the 1st and 2nd quartiles.
Major CVEs were significantly reduced by alirocumab in the baseline 3rd (HR 0.79 (95% CI 0.66, 0.95)) and 4th quartiles (HR 0.83 (95% CI 0.70, 0.98)). Similar HRs were obtained after adjusting as detailed above. When the time-weighted moving average Lp(a) change from baseline was modeled with major CVEs or non-fatal MI significant reductions of events (approximately by 16%) were seen, even after adjusting for demographic variables (age, sex, race, region), baseline Lp(a), baseline LDL-C, change in LDL-C, and clinical variables (time from acute coronary syndrome, body mass index, diabetes, smoking history).
Conclusions
An elevation of Lp(a) is currently no accepted indication for PCSK9 inhibitors. Two reasons explain this situation: (1) comparing with the effect on LDL-C concentrations, the decrease of Lp(a) under PCSK9 inhibitors is rather small—even absent in many patients, and (2) the association of the described reduction of Lp(a) levels by 20–30% with CVEs was unknown—new data on this topic appeared only recently.
In two prospective controlled intervention studies with both available PCSK9 inhibitors (evolocumab, alirocumab) it could be shown that elevated baseline Lp(a) levels represent an atherogenic risk factor, independently of baseline LDL-C concentrations.
It has to be remembered that Lp(a) levels were no inclusion criterion. Nonetheless, in the FOURIER Study approximately 33.1% of patients had a baseline concentration higher than 120 nmol/l (or approximately 50 mg/dl) which is believed to be the 80th percentile in a general patient population [15].
The lowering of Lp(a) with the injection therapy reduced the rate of CVEs—with modeling an influence of baseline values or PCSK9 inhibitor-induced changes of LDL-C or other factors could be excluded. The higher the baseline Lp(a) concentrations were, the higher the reduction of CVEs by the PCSK9 inhibitors was. These findings clearly put this new class of lipid-lowering drugs into another perspective. Possibly high Lp(a) levels will be taken into consideration when considering the use of these drugs in the future. Of course, this indication would be valid only in patients whose Lp(a) levels really demonstrate a decrease on this injection therapy.
The missing effect of PCSK9 inhibitors on Lp(a) concentrations in up to 30% of patients is not yet fully understood. Reasons, discussed in the literature, are the following: (1) apo(a) with a low kringle IV type 2 number may less actively bind to the LDLR, and (2) because furin-cleaved PCSK9 is somewhat less effective on binding to LDLRs compared with the intact PCSK9 form, it is possible that the balance between forms, as influenced by treatment with a PCSK9 inhibitor, also contributes to the degree of Lp(a) reduction on therapy [1].
The current therapeutic approach to improve the high-risk situation in patients with high Lp(a) levels is to optimize LDL-C below 1.8 mmol/l (70 mg/dl).
Given the proposed potentiation of the CVD risk between LDL-C and Lp(a), Verbeek et al. hypothesized in 2018 that the risk associated with elevated Lp(a) levels would largely be attenuated at lower LDL-C levels [17]. They tested this hypothesis in two large studies corresponding to a primary prevention setting: the European Prospective Investigation of Cancer (EPIC) Norfolk prospective population study and the Copenhagen City Heart Study prospective population study. At LDL-C levels, corrected for Lp(a)-derived LDL‑C, less than 2.5 mmol/l (~100 mg/dl), the risk associated with elevated Lp(a) decreases [17].
On the other hand, in statin studies where usually patients within secondary prevention were included, this situation is different. In an individual-patient data meta-analysis of statin-treated patients, patient-level data from seven randomized, placebo-controlled, statin outcomes trials were collated and harmonized to calculate HRs for CVEs, defined as fatal or non-fatal coronary heart disease, stroke, or revascularization procedures [18]. Elevated baseline and on-statin Lp(a) showed an independent approximately linear relation with cardiovascular disease risk, evident on treatment with either statin or placebo.
Statins in contrast to PCSK9 inhibitors do not decrease Lp(a) levels, but may even increase them. The parallel effect on both LDL-C and Lp(a) concentrations, as described in PCSK9 inhibitor studies, appears to be a major progress. It could be clearly shown that the optimization of both parameters reduces risk, but even the additional decrease of LDL-C represents an advantage.
The measured LDL-C also contains cholesterol that is transported with the Lp(a) particles [19]. This is a problem in patients who on PCSK9 inhibitor therapy achieved rather low LDL-C levels and who still show high Lp(a) values.
However, some patients still develop CVEs although both target levels were rather low during the course of the studies. The life-long burden with these lipoproteins could be a possible explanation; both LDL-C and Lp(a) are genetically determined.
In this context, the authors wish to emphasize that the rate of new events in high-risk patients with elevated Lp(a) concentrations on LA therapy is much lower [20, 21].
In the future, an antisense oligonucleotide against Lp(a) will be tested in a phase III study which will probably start in 2020. This study will clarify the effect of a of Lp(a) reduction by more than 90% on CVEs [22].
References
Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM et al (2018) NHLBI working group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol 71(2):177–192. https://doi.org/10.1016/j.jacc.2017.11.014
Sharma M, Redpath GM, Williams MJ, McCormick SP (2017) Recycling of apolipoprotein(a) after PlgRKT-mediated endocytosis of lipoprotein(a). Circ Res 120(7):1091–1102. https://doi.org/10.1161/CIRCRESAHA.116.310272
Schulz R, Schluter KD (2017) PCSK9 targets important for lipid metabolism. Clin Res Cardiol Suppl. https://doi.org/10.1007/s11789-017-0085-0
Reyes-Soffer G, Pavlyha M, Ngai C, Thomas T, Holleran S, Ramakrishnan R et al (2017) Effects of PCSK9 inhibition with alirocumab on lipoprotein metabolism in healthy humans. Circulation 135(4):352–362. https://doi.org/10.1161/CIRCULATIONAHA.116.025253
Tsimikas S (2017) A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol 69(6):692–711. https://doi.org/10.1016/j.jacc.2016.11.042
Tsimikas S, Brilakis ES, Miller ER, McConnell JP, Lennon RJ, Kornman KS et al (2005) Oxidized phospholipids, Lp(a) lipoprotein, and coronary artery disease. N Engl J Med 353(1):46–57. https://doi.org/10.1056/NEJMoa043175
Tavori H, Christian D, Minnier J, Plubell D, Shapiro MD, Yeang C et al (2016) PCSK9 association with lipoprotein(a). Circ Res 119(1):29–35. https://doi.org/10.1161/CIRCRESAHA.116.308811
Nave AH, Lange KS, Leonards CO, Siegerink B, Doehner W, Landmesser U et al (2015) Lipoprotein (a) as a risk factor for ischemic stroke: a meta-analysis. Atherosclerosis 242(2):496–503
Langsted A, Kamstrup PR, Nordestgaard BG (2019) High lipoprotein(a) and high risk of mortality. Eur Heart J. https://doi.org/10.1093/eurheartj/ehy902
Cao YX, Liu HH, Li S, Li JJ (2018) A Meta-analysis of the effect of PCSK9-monoclonal antibodies on circulating lipoprotein (a) levels. Am J Cardiovasc Drugs. https://doi.org/10.1007/s40256-018-0303-2
Watts GF, Chan DC, Somaratne R, Wasserman SM, Scott R, Marcovina SM et al (2018) Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur Heart J 39(27):2577–2585. https://doi.org/10.1093/eurheartj/ehy122
Stiekema LCA, Stroes ESG, Verweij SL, Kassahun H, Chen L, Wasserman SM et al (2018) Persistent arterial wall inflammation in patients with elevated lipoprotein(a) despite strong low-density lipoprotein cholesterol reduction by proprotein convertase subtilisin/kexin type 9 antibody treatment. Eur Heart J. https://doi.org/10.1093/eurheartj/ehy862
Moriarty PM, Parhofer KG, Babirak SP, Cornier MA, Duell PB, Hohenstein B et al (2016) Alirocumab in patients with heterozygous familial hypercholesterolaemia undergoing lipoprotein apheresis: the ODYSSEY ESCAPE trial. Eur Heart J 37(48):3588–3595. https://doi.org/10.1093/eurheartj/ehw388
Giugliano RP, Pedersen TR, Park JG, De Ferrari GM, Gaciong ZA, Ceska R et al (2017) Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the FOURIER trial. Lancet 390(10106):1962–1971. https://doi.org/10.1016/S0140-6736(17)32290-0
O’Donoghue ML, Fazio S, Giugliano RP, Stroes ESG, Kanevsky E, Gouni-Berthold I et al (2018) Lipoprotein(a), PCSK9 inhibition and cardiovascular risk: insights from the FOURIER trial. Circulation. https://doi.org/10.1161/CIRCULATIONAHA.118.037184
Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R et al (2018) Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med 379(22):2097–2107. https://doi.org/10.1056/NEJMoa1801174
Verbeek R, Hoogeveen RM, Langsted A, Stiekema LCA, Verweij SL, Hovingh GK et al (2018) Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur Heart J 39(27):2589–2596. https://doi.org/10.1093/eurheartj/ehy334
Willeit P, Ridker PM, Nestel PJ, Simes J, Tonkin AM, Pedersen TR et al (2018) Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: individual patient-data meta-analysis of statin outcome trials. Lancet 392(10155):1311–1320. https://doi.org/10.1016/S0140-6736(18)31652-0
Yeang C, Witztum JL, Tsimikas S (2015) ‘LDL-C’ = LDL-C + Lp(a)-C: implications of achieved ultra-low LDL-C levels in the proprotein convertase subtilisin/kexin type 9 era of potent LDL-C lowering. Curr Opin Lipidol 26(3):169–178
Leebmann J, Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D et al (2013) Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: prospective observational multicenter study. Circulation 128(24):2567–2576. https://doi.org/10.1161/CIRCULATIONAHA.113.002432
Roeseler E, Julius U, Heigl F, Spitthoever R, Heutling D, Breitenberger P et al (2016) Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol 36(9):2019–2027. https://doi.org/10.1161/ATVBAHA.116.307983
Viney NJ, van Capelleveen JC, Geary RS, Xia S, Tami JA, Yu RZ et al (2016) Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 388(10057):2239–2253. https://doi.org/10.1016/S0140-6736(16)31009-1
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
U. Julius: honoraria from Aegerion, Akcea, Amgen, Amryt, Chiesi, Sanofi, Kaneka, Diamed, Fresenius Medical Care, MSD. S. Tselmin received honoraria for lectures and consulting by Amgen, Fresenius Medical Care, Kaneka, MSD, and Sanofi-Aventis. S. Fischer: honoraria from Sanofi, Amgen, MSD, Berlin-Chemie, Abbott, Boehringer Ingelheim. U. Schatz and S. R. Bornstein declare that they have no competing interests.
Additional information
This article is part of the special issue “Lp(a) – Update 2018”
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Julius, U., Tselmin, S., Schatz, U. et al. Lipoprotein(a) and proprotein convertase subtilisin/kexin type 9 inhibitors. Clin Res Cardiol Suppl 14 (Suppl 1), 45–50 (2019). https://doi.org/10.1007/s11789-019-00099-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11789-019-00099-z