
Abstract
Several Bacillus strains are used as biocontrol agents, as they frequently have strong antagonistic effects against microbial plant pathogens. Bacillus strain SZMC 6179J, isolated from tomato rhizosphere, was previously shown to have excellent in vitro antagonistic properties against the most important fungal pathogens of tomato (Alternaria solani, Botrytis cinerea, Phytophthora infestans and Sclerotinia sclerotiorum) as well as several Fusarium species. Taxonomic investigations revealed that it is a member of the B. subtilis subsp. subtilis group and very closely related with the reference type strain B. subtilis subsp. subtilis 168. The sequenced genome of strain SZMC 6179J contains the genes responsible for the synthesis of the extracellular antibiotics surfactin, fengycin and bacilysin. Compared to strain 168, a prophage-like region is missing from the genome of SZMC 6179J, while there are 106 single nucleotide polymorphisms and 23 deletion-insertion polymorphisms. The high biocontrol potential of strain SZMC 6179J may results from a single base deletion in the sfp gene encoding the transcription factor of the surfactin and fengycin operons. Hypermutated regions reflecting short-time evolutionary processes could be detected in SZMC 6179J. The deletion-insertion polymorphism in the sfp gene and the detected hypermutations can be suggested as genetic determinants of biocontrol features in B. subtilis.
Graphical Abstract

Similar content being viewed by others


Introduction
Bacillus strains are successful biological pest control agents by competition for nutrients and the ecological niche in the rhizosphere. They also produce various antibiotics and extracellular enzymes and induce systemic resistance mechanisms in plants (Cawoy et al. 2011, 2015; Emmert and Handelsman 1999; Jourdan et al. 2009; Kloepper et al. 2004; Shoda 2000).
Complete genome sequences are also available for some biocontrol strains of the genus Bacillus (Borriss 2015). Earl et al. (2012) published full genomes of four B. subtilis strains (B. subtilis subsp. subtilis RO-NN-1 and AUSI98, B. subtilis subsp. spizizenii TU-B-10 and DV1-B-1) and the reference type strains DV1-F-3(T) and RO-H-1(T) of the two closely related species B. vallismortis and B. mojavensis, respectively. Zeigler (2011) revealed that the genome sequence of B. subtilis subsp. spizizenii W23 shares a 3.6 Mb core genome with the intensively studied model organism B. subtilis subsp. subtilis 168 (Zeigler et al. 2008), and the gene order within this core has been strongly conserved. Additionally, the W23 genome has 157 accessory (non-core) genome segments that are not found in B. subtilis subsp. subtilis 168, while its genome has 141 segments not found in strain W23. Deng et al. (2011) presented the complete genome sequence of B. subtilis strain BSn5 isolated from Amorphophallus konjac callus tissue and showing strong inhibitory activity to Pectobacterium carotovorum subsp. carotovorum, which causes Amorphophallus soft rot disease. Compared with B. subtilis subsp. subtilis strain 168, 9 DNA fragments (> 5 kb) were found to be inserted and 8 DNA fragments (> 5 kb) were lost in strain BSn5. The changes affect prophage sequences, cell wall synthesis, antibiotic synthesis, sporulation regulation, mobile elements, a restriction modification system and the major facilitator superfamily MFS, which may contribute to the endophytic nature of strain BSn5. Guo et al. (2013) found large gene clusters of the rhizobacterium B. subtilis XF-1 that are related to the nonribosomal synthesis of antimicrobial lipopeptides and polyketides. The strain was also found to possess a gene cluster involved in the synthesis of chitosanase responsible for the suppression of the pathogen Plasmodiophora brassicae. Guo et al. (2014) also reported the fully annotated genome of B. subtilis strain BAB-1, and identified the genes encoding for active antifungal compounds in this biocontrol strain which is highly efficient against tomato gray mold. Approximately 5.2% of the genome of strain BAB-1 was found to be devoted to the synthesis of antimicrobial products, including antibiotics produced by non-ribosomal peptide synthetases (NRPSs) and polyketide synthases (PKSs), lantibiotics, as well as bacillibactin. Among these products, the lipopeptides surfactin and fengycin were also found in the strain. Fengycin was identified as a major active antifungal compound in growth inhibition of Botrytis cinerea. When applied in combination with fengycin, surfactin showed synergistic actions which were confirmed by antifungal assay in vivo. Luo et al. (2015) analyzed the full genome of B. subtilis 916, a strain highly active against filamentous fungi. This strain not only coproduces the three families of well-known lipopeptides, i.e. surfactins, bacillomycin Ls (iturin family) and fengycins, but also produces a new family of lipopeptides called locillomycins. The genome of the strain contains four NRPS gene clusters (srf, bmy, fen and loc), which are responsible for the biosynthesis of surfactins, bacillomycin Ls, fengycins and locillomycins, respectively. The complete annotated genome sequence of B. subtilis SG6 antagonistic to Fusarium graminearum has also been released (Zhao et al. 2014). The distinct B. subtilis strains produce more than two dozens of structurally diverse antimicrobial compounds, and 4–5% of their genomes is related with antibiotic production (Stein 2005).
During this study we performed genome analysis to identify possible genetic determinants of biocontrol features in the case of B. subtilis SZMC 6179J, a strain shown to exert excellent in vitro antagonistic properties against tomato pathogens (Vágvölgyi et al. 2013). A preliminary taxonomic identification, extracellular enzyme and antibiotic secretion characteristics, and a wide in vitro antagonistic spectrum of strain SZMC 6179J were reported previously (Vágvölgyi et al. 2013). Strain SZMC 6179J produces surfactins and fengycins, and it is effective against Xanthomonas vesicatoria and a wide set of phytopathogenic filamentous fungi (Manczinger et al. 2011; Szekeres et al. 2013; Vágvölgyi et al. 2013).
Materials and methods
Bacterial strain and growth conditions
Strain B. subtilis SZMC 6179J was isolated from tomato rhizosphere during a previous study (Vágvölgyi et al. 2013) and deposited in the Szeged Microbiology Collection (SZMC; http://www.szmc.hu), Szeged, Hungary. The strain was maintained on yeast extract-glucose (YEG) medium (yeast extract 0.2%, glucose, 0.2%, bacto agar 2%) at 25 °C.
Genome sequencing
Strain SZMC 6179J was isolated two years before the whole genome sequencing from the rhizosphere of tomato as a single colony, possibly developing from a single spore, and maintained on YEG medium by subculturing about 50 times. As this 50-times-passed culture was subjected to full genome sequencing, the resulting reads also reflect the population genomics properties of the strain. Genome sequencing was performed by the cycled ligation sequencing on a SOLiD V4 system (Life Technologies) at Baygen (Szeged). Assembly was performed using the Genomics Workbench 4.7.2 (CLC Bio) and the Gapped SOLiD Alignment 1.2 plug-in (Omixon). Annotation was performed with the NCBI Prokaryotic Genome Annotation Pipeline (PGAAP) (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html). The 50-nucleotide-long reads were mapped to the annotated genome of the type strain B. subtilis subsp. subtilis 168 (GenBank ID: NC_000964) with Genomics Workbench 4.7.2. The resulting consensus sequence was used to upload the whole genome sequence of strain SZMC 6179J to the NCBI GenBank (ID: CP015004.1). The diagram showing the properties of the full genome of the strain was constructed by DNAplotter (http://www.sanger.ac.uk/science/tools/dnaplotter) (Carver et al. 2009).
Taxonomic investigations
The exact taxonomic position of the strain was determined by multilocus sequence typing (MLST), which was successfully used for the B. subtilis group by Kamada et al. (2015) who used internal fragments of seven genes (purH, glpF, pycA, ilvD, rpoD, tpiA and pta) suggested by the MLST database (PubMLST; http://pubmlst.org/bsubtilis/) (Jolley and Maiden 2010). In our study, the complete sequences of nine genes (the seven previously mentioned ones as well as gyrA and gyrB) were mined from the genome of SZMC 6179J and further 173 full Bacillus genomes. Sequences for each gene were individually aligned by PRANK v.140603 (Löytynoja 2014) with default settings, which was followed by concatenation of the alignments with SequenceMatrix 1.8 (Vaidya et al. 2011) and partitioning of the dataset by the nine loci. Maximum likelihood (ML) inferences were generated from the dataset with raxmlGUI 1.5b1 (Silvestro and Michalak 2012) using the executables of RAxML 8.2.7 (Stamatakis 2014) under the GTR model with gamma-distributed rate heterogeneity with 1000 thorough bootstrap replicates.
Mining of secondary metabolism-related genes and prophage cluster sequences, single nucleotide polymorphisms (SNPs) and deletion-insertion polymorphisms (DIPs)
Gene clusters of putative antimicrobials were searched for by the web-based genome mining tool antiSMASH (http://antismash.secondarymetabolites.org) (Blin et al. 2013; Medema et al. 2011; Weber et al. 2015). The presence of prophage sequences in the Bacillus genomes was analyzed with the PHAST search system (http://phast.wishartlab.com) (Zhou et al. 2011). The distribution of a specific prophage-like region in Bacillus strains was examined by BLAST (Zhang et al. 2000) and the results were visualized with the Kablammo server (Wintersinger and Wasmuth 2015). SNPs and DIPs were mined from the aligned reads with CLC Sequence Viewer v.6.5.3. and CLC Genomics Workbench 5.1.
In vitro antifungal activity testing
Confrontation tests were carried out on YEG medium. Strain B. subtilis SZMC 6179J and a series of phytopathogenic fungi from the Szeged Microbiology Collection (http://www.szmc.hu) listed in Table 4 were inoculated on the surface of agar plates with 3 cm spacing. Control plates were inoculated only with the respective phytopathogenic fungus. After 5 days of incubation, the colony radii of the phytopathogenic fungi were recorded and biocontrol index (BCI) values were calculated according to the formula: BCI = (C − T)/C × 100, where C and T are the colony radius values of the phytopathogenic fungi in the absence and presence of the bacterium, respectively (Nene and Thapliyal 1993).
Results
Main characteristics of the complete genome of Bacillus subtilis SZMC 6179J
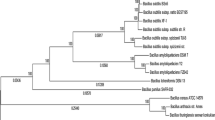
Bacillus subtilis SZMC 6179J has a single circular chromosome of 4,195,195 bp (GenBank ID: CP015004.1, Fig. 1), with a GC content of 43.6%, 4276 coding genes, 46 tRNAs, 10 rRNA loci and 13 pseudogenes (Fig. 1A). Compared to the reference genome of the type strain B. subtilis subsp. subtilis 168, large segments are missing from the genome of strain SZMC 6179J in nucleotide regions 529,444–536,858, 536,946–548,309 and 548,412–549,854. These segments are occurring within a region of the reference genome, which contains some genes of phage origin (Figs. 1B, 2), suggesting that perhaps the entire region is a prophage, or a modified prophage cluster. However, the investigation of the reference strain’s genome for the distribution of prophage sequences by PHAST (Phage Search Tool) did not reveal any prophages or prophage traces. The presence of this region was examined in other Bacillus strains by BLAST (Basic Local Alignment Search Tool) and the results were visualized with Kablammo, which revealed that besides B. subtilis subsp. subtilis 168, only 11 other strains have this prophage-like region in an intact form, while it is entirely missing from SZMC 6179J and a series of other B. subtilis strains. Strains with the full prophage-like region are present only in the “Group I” of B. subtilis subsp. subtilis (Fig. 3).

Comparison of the genomes of B. subtilis SZMC 6179J (A) and B. subtilis subsp. subtilis strain 168 (B). Intact, incomplete and questionable prophages are indicated according to the hits of PHAST searches. B also shows the prophage-like region 529,444–549,854 which is not detected by PHAST (light blue box)

The prophage-like region of B. subtilis subsp. subtilis strain 168, which is missing from B. subtilis SZMC 6179J. ydcL: phage integrase; immA: immunity anti-repressor conserved in prophages; immR: phage element transcriptional regulator; sacV: transcriptional regulator with extrachromosomal origin; ydzL, ydcO, ydcP: hypothetical proteins; ydcQ: DNA wielding protein; ydcR: replication protein, mobile element region; ydcS, ydcT, yddA-yddG: hypothetical proteins; yddH: cell wall hydrolase, mobile element region; yddL: hypothetical protein; yddJ: lipoprotein; yddK: hypothetical protein; yddM: helicase mobile element region; rapL: response regulator aspartate phosphatase; phrL: secreted regulator of the phosphatase

Maximum Likelihood phylogenetic tree of Bacillus subtilis strains constructed on the basis of nine complete gene sequences (gyrA, gyrB, purH, glpF, pycA, ilvD, rpoD, tpiA and pta) by the MLST approach. Numbers at branches indicate bootstrap values estimated by 1000 thorough bootstrap replicates under the GTR + Γ model with ten partitions. Strains containing the full prophage-like region (corresponding to 529,444–549,854 in B. subtilis subsp. subtilis strain 168) in an intact form are marked with filled pentagon. Strains containing the full, probably functional copy of the yqcG gene are marked with filled five pointed star. A version of the tree with a higher number of related Bacillus strains is shown in Online Resource Fig. 1
The exact taxonomic position of Bacillus subtilis SZMC 6179J
Strain SZMC 6179J was identified in a previous study (Vágvölgyi et al. 2013) as Bacillus subtilis by the sequence analysis of a fragment of the gyrA gene (Genbank accession number: JX683908) according to Reva et al. (2004). Phylogenetic analysis by the MLST approach performed with the full sequences of nine genes (gyrA, gyrB, purH, glpF, pycA, ilvD, rpoD, tpiA and pta) revealed that strain SZMC 6179J is belonging to the B. subtilis subsp. subtilis group and is closely related with the type strain B. subtilis subsp. subtilis 168 (Fig. 3, Online Resource 1).
The presence of antibiotic gene clusters in the genome of B. subtilis SZMC 6179J
Biocontrol microorganisms frequently produce distinct antibacterial and antifungal compounds. The genome of strain SZMC 6179J was analyzed for gene clusters of putative antimicrobials by the web-based genome mining tool antiSMASH (antibiotics and Secondary Metabolite Analysis Shell). The outlined clusters are presented in Table 1. We investigated the distribution of gene clusters of surfactin and fengycin in other strains of the genus Bacillus with the microbiological BLAST system at NCBI (National Center for Biotechnology Information) against full genomes. The structures of these operons are shown on Fig. 4. BLAST analyses with the surfactin and fengycin operons revealed that the full size surfactin operon is more abandoned in the genus than the fengycin operon. Strain SZMC 6179J contains both operons and in a potentially functional state. In the fengycin operon, large or complete deletions are frequent in other Bacillus strains.

The structure of surfactin (srfA) (A) and plipastatin (= fengycin) (B) operons of B. subtilis subsp. subtilis str. 168. The small comS gene within the srfAB gene is a regulator gene responsible for genetical competence
SNPs and DIPs in comparison to the reference genome
CLC Sequence Viewer v.6.5.3 detected 106 SNPs in the genome of SZMC 6179J in comparison to the reference genome of B. subtilis subsp. subtilis strain 168. For the full list of SNPs see Online Resource 2. The data of Table 2 suggest that important functions (active genes for antibiotics and extracellular enzymes) necessary for biocontrol were not lost due to these SNPs. In the genome of strain SZMC 6179J, 23 deletion/insertion type variants (DIPs) were found; 9 out of which could be allocated to structural genes (Online Resource 3).
In strain SZMC 6179J a single nucleotide deletion in position 407,533 causes a frameshift in the sfp gene - encoding a phosphopantetheine-transferase-compared to strain 168 in a way that it results in a normal size Sfp. The size of the extracted gene sequence is 675 bp. After translation, a BLASTP search performed at NCBI showed that the deletion resulted in a complete amino acid sequence identical with that of the following 10 Bacillus Sfp proteins: B. subtilis subsp. subtilis str. NCIB 3610 (ABV89947.1), B. subtilis subsp. subtilis str. NCIB 3610 (ABV89950.1), B. subtilis MB73/2 (EME05049.1), B. subtilis subsp. subtilis 6051-HGW (AGG59700.1), B. subtilis subsp. subtilis (KFH30033.1), B. subtilis subsp. subtilis (KFH34853.1), B. subtilis KCTC 1028 (AKC45888.1), Bacillus sp. LM 4 − 2 (AKE22172.1), B. subtilis (KNB76119.1) and B. murimartini (KON99158.1).
Mutation hotspots in the genome of B. subtilis SZMC 6179J
The strain was isolated 2 years before the whole genome sequencing, and during this time it was maintained on YEG medium by subculturing about 50 times. For genome sequencing, the DNA was extracted from this culture and not from a single cell-based culture. In this way the obtained reads reflect the genetical structure of the cell population developed through 2 years. The applied Next-Generation Sequencing method (SOLiD) produces 50-nucleotide-long reads, the number of which was 6 531 607 in the case of strain SZMC 6179J. These were aligned to the reference genome NC_000964. Considering the genome size of strain SZMC 6179J, the average coverage level was 77.84. The aligned, mapped reads were scanned for SNPs in the reads with the software CLC Genomics Workbench 5.1. at two distinct sensitivity levels (5% and 35%) to explore regions of hypermutation (e.g., a 5% sensitivity SNP scan shows the variances at nucleotide positions occurring with at least 5% frequency in the overlapping read sequences).
Our scan results showed that SNPs are not equally distributed on the chromosome in the genome population from the SZMC 6179J culture (Fig. 5). The most allelic variants within the cell population of strain SZMC 6179J could be found in the yqcG gene, which encodes for a toxic ribonuclease. Out of the 25 SNPs detected by the 35% sensitivity scan, only 5 result in amino acid changes, thus it is quite interesting, why the other 20 SNPs are so frequent in this gene, and what can possibly be the selection advantage provided by the SNPs present in this gene. The distribution of SNPs within the yqcG gene proved to be non-random, showing three hotspots within the N-terminal region (Fig. 6), which result in frequent amino acid changes in the sequence of the toxin (Table 3). The abundance of the yqcG gene in the Bacillus genus was also investigated by nucleotide BLAST against full genomes. The full gene is present only in some strains in the genus, mainly in the near relatives of B. subtilis subsp. subtilis str. 168 (Fig. 3). It is very interesting that a few, more distant relatives of strain 168 (e.g. strains ge28, HJ0-6 or BSn5; Fig. 3) also contain the full gene without any SNPs, suggesting the possibility of horizontal gene transfer of yqcG within the B. subtilis group.

Distribution of SNPs in the SZMC 6179J cell population. A Distribution of SNPs in the full genome. B Frequency of SNPs in intergenic regions and in distinct genes in the genome population of B. subtilis strain SZMC 6179J as revealed by SNP scans at two distinct sensitivities (5% and 35%) in the aligned reads produced by SOLiD® Next-Generation Sequencing. Only those genes are indicated which contain at least two SNPs at 5% sensitivity scan

The frequency and distribution by positions of SNPs within the yqcG gene in the cell population of the SZMC 6179J strain, as revealed by three distinct scans in the aligned reads with different sensitivity (5, 20 and 35%). The symbols on the x axis show the positions of the start and stop codons of the yqcG gene
Besides yqcG, the cwlA gene contains the most SNPs, while the number of SNPs is also outstanding in the yobL, yqxH and xlyB genes.
In vitro antifungal activities of Bacillus subtilis SZMC 6179J
Strain Bacillus subtilis SZMC 6179J showed the strongest inhibition effect against Armillaria ostoyae, followed by Botrytis cinerea, Bipolaris bicolor and Armillaria gallica (Table 4). Furthermore, the observed BCI values of strain SZMC 6179J were also above 30% against the tested Armillaria mellea, Alternaria alternata, Colletotrichum gloeosporioides, Curvularia spicifera, Fusarium culmorum, Phoma cucurbitacearum and Sclerotinia sclerotiorum strains.
Discussion
Fully annotated genomes of Bacillus strains with biocontrol capabilities are important tools to understand their properties, complexity, plasticity and evolution. The genome of SZMC 6179J is closely related with the reference type strain B. subtilis subsp. subtilis str. 168—a tryptophan-requiring auxotrophic strain widely used in academic research, which was isolated from B. subtilis subsp. subtilis Marburg after X-ray mutagenesis (Burkholder and Giles 1947; Zeigler et al. 2008)—the main difference between them is the lack of a prophage gene set in SZMC 6179J, as well as 106 SNPs and 23 smaller size DIPs.
The prophage-like region present in the reference genome of the type strain B. subtilis subsp. subtilis 168 but missing from the genome of strain SZMC 6179J contains many open reading frames (ORFs) with unknown function, therefore, it may be assumed that this part of the genome could be a very ancient prophage region, or traces of a phage which is not included in the recent phage sequence databases. In the publication about the full genome of B. subtilis subsp. subtilis strain 168 (Kunst et al. 1997), this region is designated as a suspected prophage-like region due to some phage elements and high AT content.
Out of the 106 SNPs detected, 18 are located in intergenic regions. Some of these SNPs might have strong influence on the regulation networks of the bacterium, thereby affecting its competitive abilities and the intensity of antibiotic secretion. From the remaining SNPs, 46 result in base substitutions within coding regions of genes, but without any amino acid changes in their resulting protein products. Finally, 42 SNPs result in amino acid changes in their corresponding gene product. Sixteen of these are changes of neutral to basic, basic to neutral, neutral to acidic, acidic to neutral, small to large or large to small amino acid, which may influence the function of the given protein (Table 1). The depsipeptide gene clusters enabling the production of surfactins and fengycins are present in the genome of B. subtilis SZMC 6179J. Although the presence of a given gene cluster in the genome does not necessarily mean the synthesis of the corresponding antibiotic, the effective production of two antibiotics very important for antimicrobial effectiveness, the antibacterial surfactin and the antifungal fengycin, were proved earlier in the case of strain SZMC 6179J (Bóka et al. 2016; Manczinger et al. 2011; Vágvölgyi et al. 2013). The explored SNPs and DIPs do not disturb the efficient expression of these gene clusters, as the fengycins and surfactins are produced by the strain. The production of surfactins is mediated by the srfA operon in B. subtilis, which is consisting of four genes, srfA-A, srfA-B, srfA-C and srfA-D. The plipastatin (= fengycin) operon (pps) consists of five genes: ppsA, ppsB, ppsC, ppsD and ppsE (Fig. 4). These two operons encode the nonribosomal peptide synthetase (NRPS) subunits which catalyze the incorporation of amino acids into surfactin and plipastatin (Marahiel et al. 1997; Peypoux et al.1999). The genetic locus sfp, encoding a phosphopantetheine-transferase, is obligately necessary for lipopeptide production, as it converts the NRPS from the inactive apo form to the active holo form (Nakano et al. 1988; Quadri et al. 1998). The reference strain B. subtilis subsp. subtilis 168 contains the surfactin (srf) and plipastatin (pps) operons but is unable to produce these lipopeptides due to the lack of a functional sfp gene (Nakano et al. 1988). Although the sequence of the gene is complete, it contains an internal stop codon resulting in a truncated protein product, consequently an inactive phosphopantetheine transferase is produced, which also seems to be associated with the lack of antifungal properties in the case of this strain (Couette et al. 2010). When the wrong sfp was changed for a correct copy from a surfactin-producer strain of B. subtilis, both surfactin and fengycin production were intensively expressed in the transgenic line of strain 168 (Coutte et al. 2010). Our study revealed that a single base deletion in the sfp gene enables strain SZMC 6179J to produce active phosphopantetheine transferase and allows the production of the lipopeptides fengycin and surfactin, which was previously proved by TLC and HPLC investigations (Bóka et al. 2016; Vágvölgyi et al. 2013). This single base deletion in the sfp gene also makes strain SZMC 6179J a potential biocontrol candidate with good antifungal properties (Table 4).
Investigation of SNPs also reflects the variability within the cell population at genome sequence level, shows the regions of hypermutation in the genome where the mutations occur with an elevated frequency, and is suitable for the investigation of short-time evolution events (Brown et al. 2011; Waters et al. 2015). It has been suggested that the local hypermutation phenomenon in B. subtilis is in connection with the transcription-associated stationary-phase mutagenesis, which is in relation with the high expression level of the target gene and Mfd, the transcription repair coupling factor (Pybus et al. 2010; Robleto et al. 2012). Possibly this mechanism works in the affected genes of strain SZMC 6179J, at least we do not know about any other system which could produce such a high frequency of local hypermutations in B. subtilis. Regions of hypermutation detected in the genome of B. subtilis strain SZMC 6179J suggest that the fastest evolutionary events happen in genes important for competition processes and cell wall lysis. The mostly affected gene in strain SZMC 6179J was found to be the yqcG gene encoding for a toxic ribonuclease, which is related with the successful competition through a contact-dependent growth inhibition (CDI) of other members from the genus Bacillus. The product of this gene is the toxic component of a toxin-antitoxin (TA) module. The C-terminus (residues 379–531) has RNase activity and inhibits growth upon expression in E. coli. In vitro RNase activity and in vivo growth inhibition are neutralized by cognate antitoxin YqcF, but not by antitoxins specific to other toxins with the LXG toxin domain (Holberger et al. 2012). The N-terminal region of the toxin is responsible for the secretion and delivery of the toxin into the target competitor bacterial cells. It is supposed that this N-terminal sequence binds specifically to some membrane proteins of the target cell and these proteins are exploited for toxin entry into the cytoplasm (Willett et al. 2015). So if the bacterium population secretes a higher N-terminal sequence variety of the toxin YqcG, it will be able to suppress a higher number of distinct competitor bacteria (with distinct membrane protein surfaces). This perhaps might explain why the sequence variability is so high in this gene in the cell population of strain SZMC 6179J (Fig. 6). On the other hand, there is not even a single SNP variation in the C-terminal region where the RNase domain is located.
Further genes in strain SZMC 6179J affected by hypermutations included cwlA, yobL, yqxH and xlyB. The cwlA gene encodes for an N-acetylmuramyl-L-alanine amidase (Kuroda et al. 1991) with a key role in the cell wall autolysis during the sporulation processes. The YobL-CT, YxiD-CT and YqcG-CT domains from B. subtilis 168 have cytotoxic RNase activities, which are neutralized by the binding of cognate YobK, YxxD and YqcF antitoxin proteins, respectively. So the product of the yobL gene has functions like the intensively changing yqcG gene and is also important in the CDI systems which could be utilized by the strain during the successful habitat-occupying competition processes (Holberger et al. 2012). The product of yqxH is the toxic protein holin of bacteriophage origin. The holin homologue YqxH is encoded by a prophage-like sequence. Such holins can form pores in the membrane, through which the lytic enzymes of bacteriophages, which usually lack a signal peptide, gain access to the cell wall (Young and Bläsi 1995). Similarly to cwlA, xlyB is also an autolysin gene of prophage origin and its product is also an N-acetylmuramoyl-L-alanine amidase (Krogh et al. 1998). Previous studies could also show quick evolutionary events in B. subtilis strains, however, other genes were affected (Brown et al. 2011; Waters et al. 2015).
Data Availability
The full genome sequence of Bacillus subtilis SZMC 6179J was made available in the GenBank database (Accession No: CP015004.1).
References
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Tilmann W (2013) antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41:W204–W212. https://doi.org/10.1093/nar/gkt449
Bóka B, Manczinger L, Kecskeméti A, Chandrasekaran M, Kadaikunnan S, Alharbi NS, Vágvölgyi C, Szekeres A (2016) Ion trap mass spectrometry of surfactins produced by Bacillus subtilis SZMC 6179J reveals novel fragmentation features of cyclic lipopeptides. Rapid Commun Mass Spectrom 30:1581–1590. https://doi.org/10.1002/rcm.7592
Borriss R (2015) Bacillus, a plant beneficial bacterium. In: Lugtenberg B (ed) Principles of plant–microbe interactions. Microbes for sustainable agriculture. Springer, Berlin, pp 379–391
Brown CT, Fishwick LK, Chokshi BM, Cuff MA (2011) Whole-genome sequencing and phenotypic analysis of Bacillus subtilis mutants following evolution under conditions of relaxed selection for sporulation. Appl Environ Microbiol 77:6867–6877. https://doi.org/10.1128/AEM.05272-11
Burkholder PR, Giles NH (1947) Induced biochemical mutations in Bacillus subtilis. Am J Bot 34:345–348
Carver T, Thomson N, Bleasby A, Berriman M, Parkhill J (2009) DNA Plotter: circular and linear interactive genome visualization. Bioinformatics 25:119–120. https://doi.org/10.1093/bioinformatics/btn578
Cawoy H, Bettiol W, Fickers P, Ongena M (2011) Bacillus-based biological control of plant diseases. In: Stoytcheva M (ed) Pesticides in the modern world: pesticides use and management. InTech, Rijeka, pp 274–302
Cawoy H, Debois D, Franzil L, De Pauw E (2015) Lipopeptides as main ingredients for inhibition of fungal phytopathogens by Bacillus subtilis/amyloliquefaciens. Microb Biotechnol 8:281–295. https://doi.org/10.1111/1751-7915.12238
Coutte F, Leclére V, Béchet M, Guez JS (2010) Effect of pps disruption and constitutive expression of srfA on surfactin productivity, spreading and antagonistic properties of Bacillus subtilis 168 derivatives. J Appl Microbiol 109:480–491. https://doi.org/10.1111/j.1365-2672.2010.04683.x
Deng Y, Zhu Y, Wang P, Zhu L (2011) Complete genome sequence of Bacillus subtilis BSn5, an endophytic bacterium of Amorphophallus konjac with antimicrobial activity for the plant pathogen Erwinia carotovora subsp. Carotovora J Bacteriol 193:2070–2071. https://doi.org/10.1128/JB.00129-11
Earl AM, Eppinger M, Fricke WF, Rosovitz MJ (2012) Whole-genome sequences of Bacillus subtilis and close relatives. J Bacteriol 194:2378–2379. https://doi.org/10.1128/JB.05675-11
Emmert EAB, Handelsman J (1999) Biocontrol of plant disease: a (Gram-) positive perspective. FEMS Microbiol Lett 171:1–9. https://doi.org/10.1111/j.1574-6968.1999.tb13405.x
Guo S, Mao Z, Wu Y, Hao K (2013) Genome sequencing of Bacillus subtilis strain XF-1 with high efficiency in the suppression of Plasmodiophora brassicae. Genome Announc 1:e0006613. https://doi.org/10.1128/genomeA.00066-13
Guo Q, Li S, Lu X, Zhang X (2014) Complete genome sequence of Bacillus subtilis BAB-1, a biocontrol agent for suppression of tomato gray mold. Genome Announc 2(4):e00744–e00714. https://doi.org/10.1128/genomeA.00744-14
Holberger LE, Garza-Sánchez F, Lamoureux J, Low DA (2012) A novel family of toxin/antitoxin proteins in Bacillus species. FEBS Lett 586:132–136. https://doi.org/10.1016/j.febslet.2011.12.020
Jolley KA, Maiden M (2010) BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinform 11:595. https://doi.org/10.1186/1471-2105-11-595
Jourdan E, Henry G, Duby F, Dommes J (2009) Insights into the defense-related events occurring in plant cells following perception of surfactin-type lipopeptide from Bacillus subtilis. Mol Plant Microbe Interact 22:456–468. https://doi.org/10.1094/MPMI-22-4-0456
Kamada M, Hase S, Fujii K, Miyake M, Sato K, Kimura K, Sakakibara Y (2015) Whole-genome sequencing and comparative genome analysis of Bacillus subtilis strains isolated from non-salted fermented soybean foods. PLoS ONE 10:e0141369. https://doi.org/10.1371/journal.pone.0141369
Kloepper JW, Ryu CM, Zhang S (2004) Induced systemic resistance and promotion of plant growth by Bacillus spp. Phytopathology 94:1259–1266. https://doi.org/10.1094/PHYTO.2004.94.11.1259
Krogh S, Jørgensen ST, Devine KM (1998) Lysis genes of the Bacillus subtilis defective prophage PBSX. J Bacteriol 180:2110–2117
Kunst F, Ogasawara N, Moszer I et al (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256. https://doi.org/10.1038/36786
Kuroda A, Imazeki M, Sekiguchi J (1991) Purification and characterization of a cell wall hydrolase encoded by the cwlA gene of Bacillus subtilis. FEMS Microbiol Lett 65:9–13. https://doi.org/10.1111/j.1574-6968.1991.tb04703.x
Löytynoja A (2014) Phylogeny-aware alignment with PRANK. Meth Mol Biol 1079:155–170. https://doi.org/10.1007/978-1-62703-646-7_10
Luo C, Liu X, Zhou H, Wang X (2015) Nonribosomal peptide synthase gene clusters for lipopeptide biosynthesis in Bacillus subtilis 916 and their phenotypic functions. Appl Environ Microbiol 81:422–431. https://doi.org/10.1128/AEM.02921-14
Manczinger L, Bóka B, Vörös M, Sajben E (2011) Influence of culture conditions on the antibiotic production of antagonistic Bacillus strains isolated from tomato rhizosphere. Agrár- és Vidékfejlesztési Szemle 6:S408–S412
Marahiel MA, Stachelhaus T, Mootz HD (1997) Modular peptide synthetases involved in nonribosomal peptide synthesis. Chem Rev 97:2651–2674. https://doi.org/10.1021/cr960029e
Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R (2011) antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:339–346. https://doi.org/10.1093/nar/gkr466
Nakano MM, Marahiel MA, Zuber P (1988) Identification of a genetic locus required for biosynthesis of the lipopeptide antibiotic surfactin in Bacillus subtilis. J Bacteriol 170:5662–5668. https://doi.org/10.1128/jb.170.12.5662-5668.1988
Nene YL, Thapliyal PN (1993) Fungicides in Plant Disease Control, Ed. 3. International Science Publisher, New York
Peypoux F, Bonmatin JM, Wallach J (1999) Recent trends in the biochemistry of surfactin. Appl Microbiol Biotechnol 51:553–563. https://doi.org/10.1007/s002530051432
Pybus C, Pedraza-Reyes M, Ross CA, Martin H (2010) Transcription-associated mutation in Bacillus subtilis cells under stress. J Bacteriol 192:3321–3328. https://doi.org/10.1128/JB.00354-10
Quadri LE, Weinreb PH, Lei M, Nakano MM (1998) Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 37:1585–1595. https://doi.org/10.1021/bi9719861
Reva ON, Dixelius C, Meijer J, Priest FG (2004) Taxonomic characterization and plant colonizing abilities of some bacteria related to Bacillus amyloliquefaciens and Bacillus subtilis. FEMS Microbiol Ecol 48:249–259. https://doi.org/10.1016/j.femsec.2004.02.003
Robleto EA, Martin HA, Pedraza-Reyes M (2012) Mfd and transcriptional derepression cause genetic diversity in Bacillus subtilis. Front Biosci 4:1246–1254. https://doi.org/10.2741/E455
Shoda M (2000) Bacterial control of plant diseases. J Biosci Bioeng 89:515–521. https://doi.org/10.1016/S1389-1723(00)80049-3
Silvestro D, Michalak I (2012) RaxmlGUI: a graphical front-end for RAxML. Org Divers Evol 12:335–337. https://doi.org/10.1007/s13127-011-0056-0
Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis andpost-analysis of large phylogenies. Bioinformatics 30:1312–1313. https://doi.org/10.1093/bioinformatics/btu033
Stein T (2005) Bacillus subtilis antibiotics: structures, syntheses and specific functions. Mol Microbiol 56:845–857. https://doi.org/10.1111/j.1365-2958.2005.04587.x
Szekeres A, Bóka B, Bencsik O, Sajben E (2013) Identification of surfactin homologues from a Bacillus subtilis strain using ion-trap mass spectrometry. Acta Microbiol Immunol Hung 60(S):88
Vágvölgyi C, Sajben-Nagy E, Bóka B, Vörös M, Berki A, Palágyi A, Krisch J, Škrbić B, Đurišić-Mladenović N, Manczinger L (2013) Isolation and characterization of antagonistic Bacillus strains capable to degrade ethylenethiourea. Curr Microbiol 66:243–250. https://doi.org/10.1007/s00284-012-0263-8
Vaidya G, Lohman DJ, Meier R (2011) SequenceMatrix: concatenation software for the fast assembly of multi-gene datasets with character set and codon information. Cladistics 27:171–180. https://doi.org/10.1111/j.1096-0031.2010.00329.x
Waters SM, Zeigler DR, Nicholson WL (2015) Experimental evolution of enhanced growth by Bacillus subtilis at low atmospheric pressure: genomic changes revealed by whole-genome sequencing. Appl Environ Microbiol 81:7525–7532. https://doi.org/10.1128/AEM.01690-15
Weber T, Blin K, Duddela S et al (2015) antiSMASH 3.0: a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res 43:237–243. https://doi.org/10.1093/nar/gkv437
Willett JLE, Gucinski GC, Fatherree JP, Low DA (2015) Contact-dependent growth inhibition toxins exploit multiple independent cell-entry pathways. Proc Natl Acad Sci USA 112:11341–11346. https://doi.org/10.1073/pnas.1512124112
Wintersinger JA, Wasmuth JD (2015) Kablammo: an interactive, web-based BLAST results visualizer. Bioinformatics 31:1305–1306. https://doi.org/10.1093/bioinformatics/btu808
Young R, Bläsi U (1995) Holins: form and function in bacteriophage lysis. FEMS Microbiol Rev 17:191–205. https://doi.org/10.1111/j.1574-6976.1995.tb00202.x
Zeigler DR (2011) The genome sequence of Bacillus subtilis subsp. spizizenii W23: insights into speciation within the B. subtilis complex and into the history of B. subtilis genetics. Microbiology 157:2033–2041. https://doi.org/10.1099/mic.0.048520-0
Zeigler DR, Prágai Z, Rodriguez S, Chevreux B, Muffler A, Albert T, Bai R, Wyss M, Perkins JB (2008) The origins of 168, W23, and other Bacillus subtilis legacy strains. J Bacteriol 190:6983–6995. https://doi.org/10.1128/JB.00722-08
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7:203–214. https://doi.org/10.1089/10665270050081478
Zhao Y, Selvaraj JN, Xing F, Zhou L (2014) Antagonistic action of Bacillus subtilis strain SG6 on Fusarium graminearum. PLoS One 9(3):e92486. https://doi.org/10.1371/journal.pone.0092486
Zhou Y, Liang Y, Lynch K, Dennis JJ, Wishart DS (2011) PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. https://doi.org/10.1093/nar/gkr485
Acknowledgements
This study was supported by grant GINOP-2.3.2-15-2016-00052 (Széchenyi 2020 Programme, Hungary). The authors extend their appreciation to the International Scientific Partnership Program (ISPP) at King Saud University for funding this research work through ISPP#0105. LK is grantee of the János Bolyai Research Scholarship (Hungarian Academy of Sciences). Open access funding provided by University of Szeged (SZTE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicting interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11274_2019_2625_MOESM1_ESM.jpg
Online Resource 1. Maximum Likelihood phylogenetic tree of Bacillus strains constructed on the basis of nine genes (gyrA, gyrB, purH, glpF, pycA, ilvD, rpoD, tpiA and pta) by the multilocus sequence typing (MLST) approach. Numbers below branches indicate bootstrap values estimated by 1000 thorough bootstrap replicates under the GTR + Γ model with ten partitions. (DOCX 63 KB)
11274_2019_2625_MOESM2_ESM.docx
Online Resource 2. Full list of SNPs by position detected at 35% sensitivity scan in the genome of strain SZMC 6179J in comparison to the reference genome B. subtilis subsp. subtilis str. 168 (NC_000964.1). (DOCX 63 KB)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
Cite this article
Bóka, B., Manczinger, L., Kocsubé, S. et al. Genome analysis of a Bacillus subtilis strain reveals genetic mutations determining biocontrol properties. World J Microbiol Biotechnol 35, 52 (2019). https://doi.org/10.1007/s11274-019-2625-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11274-019-2625-x